
CYP450:药物代谢的第一相反应.主要在肝细胞的光面内质网(微粒体)进行,此过程系 由一组混合功能氧化酶系(又称药酶)所催化促进,其中最重要的是P450和有关的辅酶 类。P450酶系包括二个重要的蛋白质组分:含铁的血红素蛋白和黄素蛋白 后者能从 NADPH将电子转移至P450底物复合体。药物与P450结合位点与血红素分子非常接近, 有利于电子的转移。药物与氧化型P450结合,此时血红素的铁为三价铁(F®),通过 NADPH还原臨的作用,将NADPH的电子转移给P450.使其还原.血红素铁成二价 (Fe2)。还原型的P450药物复合物与氧分子作用.成为含氧复合物,并接受NADPH所提 供的电子,与氧生成H,0,同时药物也被氧化,P450又成为氧化型(F®)。如此反复循 环,使药物进行第一相的代谢。 P450实际上为同 一家庭的多种异构型。迄今为止。 人类P450的基因已发现有27种,编 码多种P450。基本上分成至少4个基因族,又可进一步区分为不同亚族。其分类为 CYP1,CYP2,CYP3和CYP4,亚族的分类按英语A、B、C和阿拉伯数字1,2,3等进一步 分类。按其功能,人类的P450可分成二类。CYP1,2,3,主要代谢外源性化合物,如药 物、毒物等,有交叉的底物特异性,常可被外源性物质诱导,在讲化时程中.其保守性 差。GYP4则主要代谢内源性物质,有高度特异性,通常不能被外源性物质诱导,在进行 过程中相对保守。此类P450在类固醇、脂防酸和前列腺素代谢中起作用。在药物代谢中 起重要作用的P450。 表2-1具有代表性药物代谢CYP1,CYP2和CYP3亚家族 P450亚族 代谢的底物(药物】 CYP1A2 氧阿米替林,咖啡因,氣哌啶醇,茶碱,他克林,西咪替丁 CYP2B6 环磷酰胺 卡马西平,环磷酰胺,地西泮,布洛芬,奈普生,奥美拉唑,苯 CYP2C 妥英,普奈洛尔,甲苯磺西脲 CYP2D6 异喹胍,大多数B受体拮抗剂,氧阿米替林,氯丙嗪,可待因,右 美沙芬,恩卡尼,氟哌啶醇,去甲替林,维拉帕米 CYP2E 对乙酰氨基酚,乙醇,氟烷 胺碘酮,卡马西平,西沙必利,可卡因,皮质醇,环孢素,氨苯 砜,地塞米松,地尔硫草,红霉素,丙米嗪,利多卡因,洛伐他 CYP3A 汀,硝苯地平,孕酮,他克莫司,他莫昔芬,孝丸酮丙戊酸 盐,维拉帕米,长春新碱,华法令
CYP450:药物代谢的第一相反应,主要在肝细胞的光面内质网(微粒体)进行,此过程系 由一组混合功能氧化酶系(又称药酶)所催化促进,其中最重要的是 P450 和有关的辅酶 类。P450 酶系包括二个重要的蛋白质组分:含铁的血红素蛋白和黄素蛋白,后者能从 NADPH 将电子转移至 P450 底物复合体。药物与 P450 结合位点与血红素分子非常接近, 有利于电子的转移。药物与氧化型 P450 结合,此时血红素的铁为三价铁(Fe3+),通过 NADPH 还原酶的作用,将 NADPH 的电子转移给 P450,使其还原,血红素铁成二价 (Fe2+)。还原型的 P450 药物复合物与氧分子作用,成为含氧复合物,并接受 NADPH 所提 供的电子,与氧生成 H2O,同时药物也被氧化,P450 又成为氧化型(Fe3+)。如此反复循 环,使药物进行第一相的代谢。 P450 实际上为同一家庭的多种异构型。迄今为止,人类 P450 的基因已发现有 27 种,编 码多种 P450。基本上分成至少 4 个基因族,又可进一步区分为不同亚族。其分类为 CYP1,CYP2,CYP3 和 CYP4,亚族的分类按英语 A、B、C 和阿拉伯数字 1,2,3 等进一步 分类。按其功能,人类的 P450 可分成二类。CYP1,2,3,主要代谢外源性化合物,如药 物、毒物等,有交叉的底物特异性,常可被外源性物质诱导,在进化过程中,其保守性 差。GYP4 则主要代谢内源性物质,有高度特异性,通常不能被外源性物质诱导,在进行 过程中相对保守。此类 P450 在类固醇、脂肪酸和前列腺素代谢中起作用。在药物代谢中 起重要作用的 P450。 表 2-1 具有代表性药物代谢 CYP1,CYP2 和 CYP3 亚家族 P450 亚族 代谢的底物(药物) CYP1A2 氧阿米替林,咖啡因,氟哌啶醇,茶碱,他克林,西咪替丁 CYP2B6 环磷酰胺 CYP2C 卡马西平,环磷酰胺,地西泮,布洛芬,奈普生,奥美拉唑,苯 妥英,普奈洛尔,甲苯磺西脲 CYP2D6 异喹胍,大多数β受体拮抗剂,氧阿米替林,氯丙嗪,可待因,右 美沙芬,恩卡尼,氟哌啶醇,去甲替林,维拉帕米 CYP2E 对乙酰氨基酚,乙醇,氟烷 CYP3A 胺碘酮,卡马西平,西沙必利,可卡因,皮质醇,环孢素,氨苯 砜,地塞米松,地尔硫草,红霉素,丙米嗪,利多卡因,洛伐他 汀,硝苯地平,孕酮,他克莫司,他莫昔芬,睾丸酮,丙戊酸 盐,维拉帕米,长春新碱,华法令
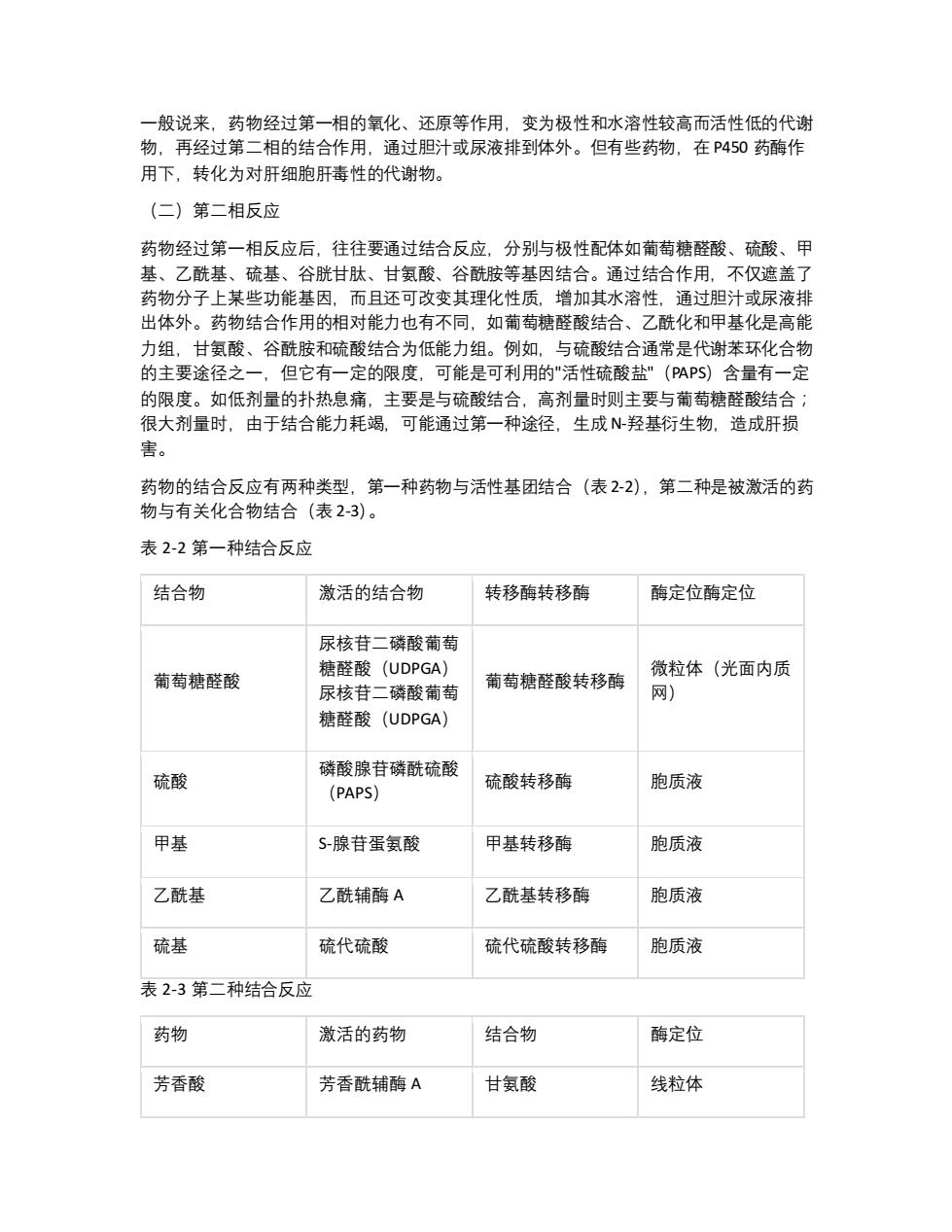
一般说来,药物经过第一相的氧化、还原等作用,变为极性和水溶性较高而活性低的代谢 物,再经过第二相的结合作用,通过胆汁或尿液排到体外。但有些药物,在P450药酶作 用下,转化为对肝细胞肝毒性的代谢物。 (二)第二相反应 药物经过第一相反应后,往往要通过结合反应,分别与极性配体如葡萄糖醛酸、硫酸、甲 基、乙酰基、硫基、谷胱甘肽、甘氨酸、谷酰胺等基因结合。通过结合作用,不仅遮盖了 药物分子上某些功能基因,而且还可改变其理化性质,增加其水溶性,通过胆汁或尿液排 出体外。药物结合作用的相对能力也有不同,如葡萄糖醛酸结合、乙酰化和甲基化是高能 力组,甘氨酸、谷酰胺和硫酸结合为低能力组。例如,与硫酸结合通常是代谢苯环化合物 的主要途径之 但它有一定的限度,可能是可利用的"活性硫酸盐”(PAPS)含量有一定 的限度。如低剂量的扑热息痛,主要是与硫酸结合,高剂量时则主要与葡萄糖醛酸结合: 很大剂量时,由于结合能力耗竭。可能通过第 种途径,生成N羟基衍生物,造成肝损 害。 药物的结合反应有两种类型,第一种药物与活性基团结合(表2-2),第二种是被激活的药 物与有关化合物结合(表2-3)。 表2-2第一种结合反应 结合物 激活的结合物 转移酶转移酶 酶定位酶定位 尿核苷二磷酸葡萄 葡萄糖醛酸 糖醛酸(UDPGA) 葡萄糖醛酸转移酶 微粒体(光面内质 尿核苷二磷酸葡萄 网) 糖醛酸(UDPGA) 硫酸 磷酸腺苷磷酰硫酸 硫酸转移酶 胞质液 (PAPS) 甲基 S腺苷蛋氨酸 甲基转移酶 胞质液 乙酰基 乙酰辅酶A 乙酰基转移酶 胞质液 硫基 硫代硫酸 硫代硫酸转移酶 胞质液 表2-3第二种结合反应 药物 激活的药物 结合物 酶定位 芳香酸 芳香酰辅酶A 甘氨酸 线粒体
一般说来,药物经过第一相的氧化、还原等作用,变为极性和水溶性较高而活性低的代谢 物,再经过第二相的结合作用,通过胆汁或尿液排到体外。但有些药物,在 P450 药酶作 用下,转化为对肝细胞肝毒性的代谢物。 (二)第二相反应 药物经过第一相反应后,往往要通过结合反应,分别与极性配体如葡萄糖醛酸、硫酸、甲 基、乙酰基、硫基、谷胱甘肽、甘氨酸、谷酰胺等基因结合。通过结合作用,不仅遮盖了 药物分子上某些功能基因,而且还可改变其理化性质,增加其水溶性,通过胆汁或尿液排 出体外。药物结合作用的相对能力也有不同,如葡萄糖醛酸结合、乙酰化和甲基化是高能 力组,甘氨酸、谷酰胺和硫酸结合为低能力组。例如,与硫酸结合通常是代谢苯环化合物 的主要途径之一,但它有一定的限度,可能是可利用的"活性硫酸盐"(PAPS)含量有一定 的限度。如低剂量的扑热息痛,主要是与硫酸结合,高剂量时则主要与葡萄糖醛酸结合; 很大剂量时,由于结合能力耗竭,可能通过第一种途径,生成 N-羟基衍生物,造成肝损 害。 药物的结合反应有两种类型,第一种药物与活性基团结合(表 2-2),第二种是被激活的药 物与有关化合物结合(表 2-3)。 表 2-2 第一种结合反应 结合物 激活的结合物 转移酶转移酶 酶定位酶定位 葡萄糖醛酸 尿核苷二磷酸葡萄 糖醛酸(UDPGA) 尿核苷二磷酸葡萄 糖醛酸(UDPGA) 葡萄糖醛酸转移酶 微粒体(光面内质 网) 硫酸 磷酸腺苷磷酰硫酸 (PAPS) 硫酸转移酶 胞质液 甲基 S-腺苷蛋氨酸 甲基转移酶 胞质液 乙酰基 乙酰辅酶 A 乙酰基转移酶 胞质液 硫基 硫代硫酸 硫代硫酸转移酶 胞质液 表 2-3 第二种结合反应 药物 激活的药物 结合物 酶定位 芳香酸 芳香酰辅酶 A 甘氨酸 线粒体

芳香基乙酸 芳香基乙酰辅酶A 谷氨酰 胞质液 芳香环化合物 环氧化合物 谷胱甘肽 胞质液 第一相的P450酶系与第二相结合作用酶系的分布、 功能和可诱导性均有差别,反映了这 二类生物转化和解毒作用的不同生物学意义。谷胱甘肽(GSH)在结合和解毒作用中起着 十分重要的作用,它能与亲电子基、氧基作用,防止肝细胞的损害。 二、影响药物代谢的因素 (一)药物代谢的遗传多态性 由于肝脏药酶系特别是P450的遗传多态性,以致造成药物代谢的个体差异,这影响了药 物的药理作用、不良反应和致癌的易感性等。 对某些药代谢的缺陷者称为:弱代谢者 (poor metabolizer)或PM-表型1.而强代谢者(extensive metabolizer)款为EM-表型 在第一相中的药物代谢多态性以异喹肌和乙妥英为例,分别为P450UD6和P4502C的变 异。对异喹胍的羟化作用有遗传性缺陷的个体,在应用β-受体拮抗剂、三环类抗郁剂、某 些膜抑制抗心律紊乱药、抗高血压药和钙离子拮抗剂等,由于药物代谢的异常,使药效增 强、时间延长,容易发生不良反应。在第二相反应的药物 代谢多态性,以异烟肼和磺胺 甲嘧啶为例,可区分为乙酰化快型和慢型两种,慢型乙酰化个体长期服用肼苯达嗪和普鲁 卡因酰胺后可产生红斑狼疹综合征服异烟册后易发生周用围神经病变(表24) P4501A1, P45012是芳香碳氢化合物羟化酶,激活某些致癌原,其遗传变异与某些癌的 易患性有关。 表2-4遗传多态性与药物代谢 代谢途径 药物举例 人群中的频率 (%) 酶 C-氧化 异喹肌,金雀花碱, 右旋甲吗喃,阿片类 白种人5-10 CYP4502D6 R竖上腺受体结抗 C氧化 剂,乙妥英,甲苯巴 白种人4 CYP4502C 比士 环已巴比土,异烟 乙酰化 肼,磺胺二甲嘧啶 日本人10 N-乙酰基转移酶白 种人30-70 咖啡因 (二)药酶的诱导和抑制
芳香基乙酸 芳香基乙酰辅酶 A 谷氨酰 胞质液 芳香环化合物 环氧化合物 谷胱甘肽 胞质液 第一相的 P450 酶系与第二相结合作用酶系的分布、功能和可诱导性均有差别,反映了这 二类生物转化和解毒作用的不同生物学意义。谷胱甘肽(GSH)在结合和解毒作用中起着 十分重要的作用,它能与亲电子基、氧基作用,防止肝细胞的损害。 二、影响药物代谢的因素 (一)药物代谢的遗传多态性 由于肝脏药酶系特别是 P450 的遗传多态性,以致造成药物代谢的个体差异,这影响了药 物的药理作用、不良反应和致癌的易感性等。对某些药代谢的缺陷者称为:弱代谢者 (poor metabolizer)或 PM-表型 1,而强代谢者(extensive metabolizer)称为 EM-表型。 在第一相中的药物代谢多态性以异喹胍和乙妥英为例,分别为 P450UD6 和 P4502C 的变 异。对异喹胍的羟化作用有遗传性缺陷的个体,在应用β-受体拮抗剂、三环类抗郁剂、某 些膜抑制抗心律紊乱药、抗高血压药和钙离子拮抗剂等,由于药物代谢的异常,使药效增 强、时间延长,容易发生不良反应。在第二相反应的药物代谢多态性,以异烟肼和磺胺二 甲嘧啶为例,可区分为乙酰化快型和慢型两种,慢型乙酰化个体长期服用肼苯达嗪和普鲁 卡因酰胺后可产生红斑狼疮综合征,服异烟肼后易发生周围神经病变(表 2-4)。 P4501A1,P4501A2 是芳香碳氢化合物羟化酶,激活某些致癌原,其遗传变异与某些癌的 易患性有关。 表 2-4 遗传多态性与药物代谢 代谢途径 药物举例 人群中的频率 (%) 酶 C-氧化 异喹胍,金雀花碱, 右旋甲吗喃,阿片类 白种人 5-10 CYP4502D6 C-氧化 β-肾上腺受体拮抗 剂,乙妥英,甲苯巴 比士 白种人 4 CYP4502C 乙酰化 环已巴比土,异烟 肼,磺胺二甲嘧啶, 咖啡因 日本人 10 N-乙酰基转移酶白 种人 30-70 (二)药酶的诱导和抑制

1酶诱导作用某些亲脂性药物或外源性物质(如农药、毒物等)可使肝内药酶的合成显著 增加,从而对其它药物的代谢能力增加,称为酶的诱导。在形态学上有光面内质网增生和 肥大。目前.已知至少有200多种的药物和环境中的化学物质。具有酶诱导的作用。其 中,比较熟知的苯巴比妥、导眠能、眠尔通、保太松、苯妥英钠、利福平、灰黄需素、安 体点特舒通、666、DDT、3甲基胆蒽和3,4苯等。药酶的诱导有时可造成药物性肝损伤 或化学致癌。环境中的杀虫剂、烟草燃烧和烧烤牛肉的产物等亦能诱导P450。 2.酶抑制作用有些药物通过抑制药酶,使另一药物的代谢延迟,药物的作用加强或延长 此即酶的抑制。微粒体药酶的专一性不高,多种药物可以作为同一酶系的底物 这样可能 出现各种药物之间对酶结合部位的竞争。对药酶亲和力低的药物,不仅它本身的代谢速率 较慢,而且当存在另一种对药酶有高亲和力药物时,它对前者的竞争能力就较差。因此 一种药物或毒物受 一种酶催化时,可以影响对其它药物的作用。已经发现保太松、双香豆 素等可抑制甲磺丁脲的代谢,而增强其降血糖作用。长期服用别嘌呤醇或去甲替林可以 造成酶抑制。氯霉素可抑制甲磺丁脲、苯妥英钠、双香豆素的代谢 (三)其他 影响药物代谢的其他有关因素有年龄(新生儿、早产儿、老年)、性别、昼夜的调节、营 养状态、饿、妊娠和内分泌等。 以上这些因素可以解释为什么不同的个体药效和不良反应出现的差异。 三、肝脏对药物的排泄 除了药物的生物转化外,肝脏对药物代谢的第二个重要功能是将药物从胆汁排泄。一般来 说,分子量大于400-500的化合物,主要直接从胆汁排泄。分子量小于300的物质进入血 液,从肾脏排出。从胆汁排出的药物,大多是已经通过第一相和第二相生物转化后已形成 的结合代谢物,但也有少数未经转变或仍呈活性状态的药物。肝脏对后者的排泄能力,直 接影响到该药在血液内的浓度,利福平就是一个例子。经胆汁排入肠道的结合代谢产物 为高度水溶性,不易从肠道吸收,随同粪便一起排出体外。但有些代谢物,在肠壁或细菌 的某些水解酶(加葡猫糖醛酸苷酶)的作用下去掉结合物又成为脂溶性.可以从肠 膜吸收 进入门静脉系统,形成肠肝循”,使药物作用的时间延长。另外,在肾功能减退 时,肝脏对药物的排泄可能是一个重要的代偿手段。 四、肝脏疾病对药物代谢的影响 肝脏疾病时,除了肝脏的药酶系和结合作用的改变可以影响药物代谢外,还有其他一些重 要的因素亦影响药物代谢和血浓度,包括肝脏的有效血流量,肝细胞对药物的摄取和排 出,有效肝细胞的总数,门体血液分流,胆道畅通情况,血浆蛋白浓度和药物的吸收 等。 药物通过&U2925;脏的总消除率(包括与肝组织结合、肝脏代谢及胆汁排泄的速率),可 用药物进出肝脏的速率差表示:
1.酶诱导作用某些亲脂性药物或外源性物质(如农药、毒物等)可使肝内药酶的合成显著 增加,从而对其它药物的代谢能力增加,称为酶的诱导。在形态学上有光面内质网增生和 肥大。目前,已知至少有 200 多种的药物和环境中的化学物质,具有酶诱导的作用。其 中,比较熟知的苯巴比妥、导眠能、眠尔通、保太松、苯妥英钠、利福平、灰黄霉素、安 体点特舒通、666、DDT、3-甲基胆蒽和 3,4-苯等。药酶的诱导有时可造成药物性肝损伤 或化学致癌。环境中的杀虫剂、烟草燃烧和烧烤牛肉的产物等亦能诱导 P450。 2.酶抑制作用 有些药物通过抑制药酶,使另一药物的代谢延迟,药物的作用加强或延长, 此即酶的抑制。微粒体药酶的专一性不高,多种药物可以作为同一酶系的底物,这样可能 出现各种药物之间对酶结合部位的竞争。对药酶亲和力低的药物,不仅它本身的代谢速率 较慢,而且当存在另一种对药酶有高亲和力药物时,它对前者的竞争能力就较差。因此, 一种药物或毒物受一种酶催化时,可以影响对其它药物的作用。已经发现保太松、双香豆 素等可抑制甲磺丁脲的代谢,而增强其降血糖作用。长期服用别嘌呤醇或去甲替林,可以 造成酶抑制。氯霉素可抑制甲磺丁脲、苯妥英钠、双香豆素的代谢。 (三)其他 影响药物代谢的其他有关因素有年龄(新生儿、早产儿、老年)、性别、昼夜的调节、营 养状态、饥饿、妊娠和内分泌等。 以上这些因素可以解释为什么不同的个体药效和不良反应出现的差异。 三、肝脏对药物的排泄 除了药物的生物转化外,肝脏对药物代谢的第二个重要功能是将药物从胆汁排泄。一般来 说,分子量大于 400-500 的化合物,主要直接从胆汁排泄。分子量小于 300 的物质进入血 液,从肾脏排出。从胆汁排出的药物,大多是已经通过第一相和第二相生物转化后已形成 的结合代谢物,但也有少数未经转变或仍呈活性状态的药物。肝脏对后者的排泄能力,直 接影响到该药在血液内的浓度,利福平就是一个例子。经胆汁排入肠道的结合代谢产物, 为高度水溶性,不易从肠道吸收,随同粪便一起排出体外。但有些代谢物,在肠壁或细菌 的某些水解酶(如葡萄糖醛酸苷酶)的作用下,去掉结合物,又成为脂溶性,可以从肠黏 膜吸收,进入门静脉系统,形成"肠肝循",使药物作用的时间延长。另外,在肾功能减退 时,肝脏对药物的排泄可能是一个重要的代偿手段。 四、肝脏疾病对药物代谢的影响 肝脏疾病时,除了肝脏的药酶系和结合作用的改变可以影响药物代谢外,还有其他一些重 要的因素亦影响药物代谢和血浓度,包括肝脏的有效血流量,肝细胞对药物的摄取和排 出,有效肝细胞的总数,门-体血液分流,胆道畅通情况,血浆蛋白浓度和药物的吸收 等。 药物通过&U 2925;脏的总消除率(包括与肝组织结合、肝脏代谢及胆汁排泄的速率),可 用药物进出肝脏的速率差表示:

药物消除率-QCA-QCv,Q代表肝血流量,CA和Cv分别代表进出肝脏的血药浓度。QCA 表示药物进入肝脏的速率,QCA表示流出的速率。药物的肝脏清除速率与药物进入肝脏速 率的关系,可用肝摄取率(extractionratio,ER)表示,它是指药物从门静脉(口服途径) 通过肝脏消除的分数。肝摄取率可介于0-1之间。如ER为0.5,表示该药从门静脉进入肝 脏后有一半被消除,其余(1)通过肝脏进入大循环。最近提出肝脏消除率可更好地表 明药物在肝脏的清除与进入肝脏药物浓度的关系,它指单位时间内有多少量(m)血浆所 含的药物被肝脏所清除。 肝脏清除率(C1H)=Q×ER 肝脏对各种药物的摄取率不同,对于高摄取率的药物(ER≌1.0)肝脏的内在清除率 (C1i1)很高,血浆中的药物通过肝脏时几乎可全部被清除,药物的肝清除率几乎等于 有效肝血流量。这类药物的清除受血流量影响大,称为流速限定性药物。肝摄取率高的药 物,受血浆蛋白结合的影响较小,口服后首次通过作用非常显著。对摄取率低( 02)的药物,肝脏的内在清除率低,受到药酶和结合酶系的影响大,而受血流量的影响较 小,称为能力限定性药物。这类药物受血浆蛋白结合影响较大,其首次通过作用不明显。 由此可见,肝病时药物清除的改变很复杂,与药物本身的理化特性也有关。一般来说,药 物代谢和清除的影响,与肝病的严重程度成正比。急性肝炎时改变较轻而短暂,失代偿期 肝硬化时则较为显著。例如在肝硬化时,保太松、氨基比林、安定、利眠宁、甲磺丁脲、 氯霉素和西米替丁等的半衰期延长,肝脏的清除率降低。在慢性或严重肝病时,由于肝脏 有效血流量降低,口服给药后使 一些高ER药物的首次通过作用受阻生物利用度增加 药物清除减慢,血药浓度升高,如水杨酸类、普萘洛尔(心得安)、氯丙嗪、利他林、吗 啡、哌替定(度冷丁)等。在严重肝病时,由于大脑的G4BA、安定和吗啡受体增多或其 敏感阑值降低,即使给于正常1/2-1/3剂量也可诱发肝性脑病。 第三节药物的肝外代谢 药物进入机体后主要以两种方式消除:一种是药物直接以原型随粪便和尿液排出体外;另 一种是药物在体内经代谢后,再以代谢物的形式随粪便和尿液排出体外。药物的代谢,也 称为生物转化,是药物从体内消除的主要方式之一。药物在体内的生物转化主要有两个步 骤:第 步代谢反应称为相反应. 药物被氧化、还原或水解;第二步代谢反应称为Ⅱ相 代谢反应,药物与一些内源性的物质(如葡萄糖醛酸、甘氨酸、硫酸等)结合或经甲基化 乙酰化后排出体外。肝脏是药物代谢的主要部位,含有许多相代谢和相代谢所需的 酶。但随着生物化学和分子生物学如蛋白质分离纯化技术、免疫抗体标记及cDNA技术的 发展和应用,越来越多的药物代谢酶在肝外组织和器官中被发现,如!相反应的主要酶系 CYP450及黄素单加氧酶FM、过氧化酶系、环氧化物水合酶等;相反应的葡萄糖醛酸 转移酶、硫酸转移酶、乙酰转移酶、甲基转移酶、氨基酸结合酶等。同时由于对药物代谢 研究的不断深入,人们发现药物的代谢不仅仅发生于肝内,有些药物如吗啡、普奈洛尔、 洛西泮等可在肝外组织中代谢:有些药物如氨基比林、红霉素、环磷酰胺和啊糖胞苷等在
药物消除率=Q·CA-Q·Cv,Q 代表肝血流量,CA 和 Cv 分别代表进出肝脏的血药浓度。Q·CA 表示药物进入肝脏的速率,Q·CA 表示流出的速率。药物的肝脏清除速率与药物进入肝脏速 率的关系,可用肝摄取率(extractionratio,ER)表示,它是指药物从门静脉(口服途径) 通过肝脏消除的分数。肝摄取率可介于 0-1 之间。如 ER 为 0.5,表示该药从门静脉进入肝 脏后有一半被消除,其余(1-ER)通过肝脏进入大循环。最近提出肝脏消除率可更好地表 明药物在肝脏的清除与进入肝脏药物浓度的关系,它指单位时间内有多少量(ml)血浆所 含的药物被肝脏所清除。 肝脏清除率(C1H)=Q×ER 肝脏对各种药物的摄取率不同,对于高摄取率的药物(ER≌1.0)肝脏的内在清除率 (C1in1)很高,血浆中的药物通过肝脏时几乎可全部被清除,药物的肝清除率几乎等于 有效肝血流量。这类药物的清除受血流量影响大,称为流速限定性药物。肝摄取率高的药 物,受血浆蛋白结合的影响较小,口服后首次通过作用非常显著。对摄取率低(ER< 0.2)的药物,肝脏的内在清除率低,受到药酶和结合酶系的影响大,而受血流量的影响较 小,称为能力限定性药物。这类药物受血浆蛋白结合影响较大,其首次通过作用不明显。 由此可见,肝病时药物清除的改变很复杂,与药物本身的理化特性也有关。一般来说,药 物代谢和清除的影响,与肝病的严重程度成正比。急性肝炎时改变较轻而短暂,失代偿期 肝硬化时则较为显著。例如在肝硬化时,保太松、氨基比林、安定、利眠宁、甲磺丁脲、 氯霉素和西米替丁等的半衰期延长,肝脏的清除率降低。在慢性或严重肝病时,由于肝脏 有效血流量降低,口服给药后使一些高 ER 药物的首次通过作用受阻,生物利用度增加, 药物清除减慢,血药浓度升高,如水杨酸类、普萘洛尔(心得安)、氯丙嗪、利他林、吗 啡、哌替定(度冷丁)等。在严重肝病时,由于大脑的 GABA、安定和吗啡受体增多或其 敏感阈值降低,即使给于正常 1/2-1/3 剂量也可诱发肝性脑病。 第三节药物的肝外代谢 药物进入机体后主要以两种方式消除:一种是药物直接以原型随粪便和尿液排出体外;另 一种是药物在体内经代谢后,再以代谢物的形式随粪便和尿液排出体外。药物的代谢,也 称为生物转化,是药物从体内消除的主要方式之一。药物在体内的生物转化主要有两个步 骤:第一步代谢反应称为Ⅰ相反应,药物被氧化、还原或水解;第二步代谢反应称为Ⅱ相 代谢反应,药物与一些内源性的物质(如葡萄糖醛酸、甘氨酸、硫酸等)结合或经甲基化、 乙酰化后排出体外。肝脏是药物代谢的主要部位,含有许多Ⅰ相代谢和Ⅱ相代谢所需的 酶。但随着生物化学和分子生物学如蛋白质分离纯化技术、免疫抗体标记及 cDNA 技术的 发展和应用,越来越多的药物代谢酶在肝外组织和器官中被发现,如Ⅰ相反应的主要酶系 CYP450 及黄素单加氧酶(FMO)、过氧化酶系、环氧化物水合酶等;II 相反应的葡萄糖醛酸 转移酶、硫酸转移酶、乙酰转移酶、甲基转移酶、氨基酸结合酶等。同时由于对药物代谢 研究的不断深入,人们发现药物的代谢不仅仅发生于肝内,有些药物如吗啡、普奈洛尔、 洛西泮等可在肝外组织中代谢;有些药物如氨基比林、红霉素、环磷酰胺和阿糖胞苷等在