
动相输送、选择泵系统时应满足以下要求:1)输液压力高,早期液相色谱的进柱压为60 70kg/©m,现在液相色谱采用高速高效分离所需的小颗粒和小直径的色谱校,则要求进柱 压大到300 500kg/cm2.2)要求高压下通过性子的液流是连续而无脉动, 流动相的流面 的变动不应超过2%,输送的流量范围:分析型为0一10ml/min,制备型为50一100ml mi。3)能耐流动相的腐蚀。4)泵的密封性要好,死体积小,以便于迅速更换溶剂和梯度洗 脱。能满足上述要求的有机械恒流泵和气体恒压泵,液相色谱中最常用的是机械往复泵。恒 定的无脉冲的高压泵能保证流量恒定和准确,获得稳定的基线和保留值重现性好,沃特斯 1erS)使用的是双泵头往复式柱塞泵, 输出流 精度士0.1%,流量的准确度为士1%,最 高压力6000psi(相当于420kg/cm2),流速调节范围为0.1一9.9ml/mim,适于高精度的液 谱分析工作,该泵配有压力传感器,可从压力表显示压力,并有压力安全选择器,设定压力 极限值,压力超过时报警并自动停泵。 二递度洗时 气相色谱分析是为了改善分离和缩短分析周期 采用程序升温控制:而在液相色增中 使用梯度洗脱。所谓梯度洗脱足在 个色谱分析周期里不断改变流动相的化学组分,使一些 复杂混合物的分析能做到:1)提高分辨能力:2)峰形得到改善:3)缩短分析周期。 梯度洗脱可分为低压梯度(外梯度)和高压梯度(内梯度)。低压梯度是在常压下将不同极 性的溶剂按预先设定的比例在泵前混合,再由高压泵输入色谱桂,高压梯度装置是用高压泵 分别将两种或三种不同极性的溶剂输入混合器,经充分混合后输入色谱柱。梯度洗服装置的 溶剂混合器应具有体积小 无死区 清洗方便和混 才能获得重复的梯度曲 经梯度洗脱后的色增柱必须回复到起始的溶剂条件,然后再进行下一次分析,如果梯度变什 在10%一15%以内,直接泵入起始浓度的移动相即可。如梯度变化很大,则逐步改变溶剂 组分,不可在100%甲醇的柱子中直接泵入20%甲醉/水,压力和流速的突然变化可引起什 床的损坏。在流动相为有机溶剂/水/盐类的体系中,必须保证盐类在整个梯度程序中完全 容解,如使用0.005mol/L庚磺酸钠,它可完全溶 甲醇:水=2:8中,但不能溶于甲醇 水=8:2中,一旦盐类发生了不溶的沉淀,会损坏色谱柱,而且不能再生 (四)进样系统 把样品引进色谱锭有两个基本方法,即注射器法和进样阀法。进样过程往往会影响分离 效果,当注入样品后,由扩散而导致峰形展宽:而在高压状态下讲杆,难度较大:还要求讲 样时不影响输液系统的压力和流速,也不出现漏液等现象。目前高效液相色谱仪多采用定显 进样阀法 阀内装有环形定量管,样品溶液在常压下 靠注射器注入贮样管 并把管中的溶剂 顶出,再靠转动阀门,在保持高压不停流状态下将样品送入流路系统,其优点是进样时不受 色谱系统压力的限制,可在常压下进样,进样量由定量进样阀调整,进样时不会发生漏液或 堵塞进样口的现象,结果重现性好,话于按装自动进样器,由微处理机控制程序,可连续自 动讲几十个样品 (五)色谱柱 色谱柱是高效液谱仪的心脏部分。柱内填充填料的种类不同,性能也不同,通常采用不 锈钢直形住,内径为2一4mm,长度为15一30cm,柱子加长能改善分离,但要提高柱入口 处的压力,且加长分析时间。近年米高效填充剂的出现,实现了分离的高速和高效,经典的 液相色谱填充剂:1)颗粒大,直径一般在100m以上:2)粒度不均匀,3)设有一定形状:4 经不住高压。高效液柏色谱填充剂:1)颗粒小, 直径为10一30m,最小为3 :2)粒 度均匀:3)一般为球形:4)能在高压下工作。近年来采用直径3一5m的填抖制成 -5cr 长的高效短柱,适于快速分析。此外,有用弹性聚四氟乙烯材料制成的径向加压柱,靠径向 加压器使填料呈均匀规则排列可改善柱效,保留值的重现性好,在一般常规分析中亦可应用。 样品中如有吸附性的组分,会影响色谱柱的使用寿命,除了应在进色谱柱前抽提或过迪
动相输送、选择泵系统时应满足以下要求:1)输液压力高,早期液相色谱的进柱压为 60 一 70 kg/cm2,现在液相色谱采用高速高效分离所需的小颗粒和小直径的色谱校,则要求进柱 压大到 300 一 500 kg/cm2。2)要求高压下通过性子的液流是连续而无脉动,流动相的流量 的变动不应超过 2%,输送的流量范围:分析型为 0-10 ml/min,制备型为 50—100ml/ min。3)能耐流动相的腐蚀。4)泵的密封性要好,死体积小,以便于迅速更换溶剂和梯度洗 脱。能满足上述要求的有机械恒流泵和气体恒压泵,液相色谱中最常用的是机械往复泵。恒 定的无脉冲的高压泵能保证流量恒定和准确,获得稳定的基线和保留值重现性好,沃特斯 (Waters)使用的是双泵头往复式柱塞泵,输出流量精度±0.1%,流量的准确度为±1%,最 高压力 6000psi(相当于 420kg/cm2),流速调节范围为 0.1—9.9ml/min,适于高精度的液 谱分析工作,该泵配有压力传感器,可从压力表显示压力,并有压力安全选择器,设定压力 极限值,压力超过时报警并自动停泵。 (三)梯度洗脱 气相色谱分析是为了改善分离和缩短分析周期,采用程序升温控制;而在液相色谱中, 使用梯度洗脱。所谓梯度洗脱足在一个色谱分析周期里不断改变流动相的化学组分,使一些 复杂混合物的分析能做到:1)提高分辨能力;2)峰形得到改善;3)缩短分析周期。 梯度洗脱可分为低压梯度(外梯度)和高压梯度(内梯度)。低压梯度是在常压下将不同极 性的溶剂按预先设定的比例在泵前混合,再由高压泵输入色谱桂,高压梯度装置是用高压泵 分别将两种或三种不同极性的溶剂输入混合器,经充分混合后输入色谱柱。梯度洗服装置的 溶剂混合器应具有体积小,无死区、清洗方便和混合效率性能,才能获得重复的梯度曲线。 经梯度洗脱后的色谱柱必须回复到起始的溶剂条件,然后再进行下一次分析,如果梯度变化 在 10%一 15%以内,直接泵入起始浓度的移动相即可。如梯度变化很大,则逐步改变溶剂 组分,不可在 l00%甲醇的柱子中直接泵入 20%甲醉/水,压力和流速的突然变化可引起什 床的损坏。在流动相为有机溶剂/水/盐类的体系中,必须保证盐类在整个梯度程序中完全 溶解,如使用 0.005mol/L 庚磺酸钠,它可完全溶于甲醇∶水=2∶8 中,但不能溶于甲醇∶ 水=8∶2 中,一旦盐类发生了不溶的沉淀,会损坏色谱柱,而且不能再生。 (四)进样系统 把样品引进色谱锭有两个基本方法,即注射器法和进样阀法。进样过程往往会影响分离 效果,当注入样品后,由扩散而导致峰形展宽;而在高压状态下进杆,难度较大;还要求进 样时不影响输液系统的压力和流速,也不出现漏液等现象。目前高效液相色谱仪多采用定量 进样阀法,阀内装有环形定量管,样品溶液在常压下靠注射器注入贮样管,并把管中的溶剂 顶出,再靠转动阀门,在保持高压不停流状态下将样品送入流路系统,其优点是进样时不受 色谱系统压力的限制,可在常压下进样,进样量由定量进样阀调整,进样时不会发生漏液或 堵塞进样口的现象,结果重现性好,适于按装自动进样器,由微处理机控制程序,可连续自 动进几十个样品。 (五)色 谱 柱 色谱柱是高效液谱仪的心脏部分。柱内填充填料的种类不同,性能也不同,通常采用不 锈钢直形住,内径为 2—4mm,长度为 15—30 cm,柱子加长能改善分离,但要提高柱入口 处的压力,且加长分析时间。近年来高效填充剂的出现,实现了分离的高速和高效,经典的 液相色谱填充剂:1)颗粒大,直径一般在 l00 µm 以上;2)粒度不均匀,3)没有一定形状;4) 经不住高压。高效液柏色谱填充剂:1)颗粒小,直径为 10 一 30 µm,最小为 3—5µm;2)粒 度均匀;3)一般为球形;4)能在高压下工作。近年来采用直径 3—5µm 的填抖制成 3—5cm 长的高效短柱,适于快速分析。此外,有用弹性聚四氟乙烯材料制成的径向加压柱,靠径向 加压器使填料呈均匀规则排列可改善柱效,保留值的重现性好,在一般常规分析中亦可应用。 样品中如有吸附性的组分,会影响色谱柱的使用寿命,除了应在进色谱柱前抽提或过滤

外,最简单的办法是在分析柱前连接一根3一5cm长的保护柱,内装与色谱柱性能相同的填 料,使用0.2μm的过滤片,放在进样器和分析柱之间。保护柱可防止来自流动相和样品中 的不溶性微粒对色 ,还可避免硅胶或键合相的流 可可地 ,保护柱并不能 无限止地吸附杂质,使用时应观察检测器讯号,如有基线漂移、怪峰或反压增加等现象,说 明保护柱已被污染,应更换填料。 色谱柱的填充方法:通常有干法和匀浆法两种,填装方法取决于填料微粒的大小,直径 大于20m,可用干法填充:与填充气相色谱柱的方法相似。直径10m或5m的微粒, 不能用干法装柱,采用匀浆方法将它们填充 色谱柱中,通常有等比重匀 浆法 是选择与硅 胶比重相似的溶剂如四溴乙烷等配成浆状液:由于该溶剂毒性力 ,硅胶对溴有吸附作用, 不常用,改用二氧六烷和四氯化碳等,国内亦常用等比重匀浆法,其步嫌如下: (1)根据柱体积称取所需之载体于三角瓶中,每g裁体加3ml二氧六环和6ml四氯化碳 匀浆液。 (2)将三角瓶置于超声波清洗器中10mi 至匀浆液呈半透明状无明显结块为止 (③)将匀浆液倒入匀浆罐中,化学键合相可在匀浆上部加 些水,旋紧顶盖,打开高压 放空阀。 (4)硅胶柱用脱水的己烷做顶替液,正相键合相用己烷做顶替液,反相键合相或离子交 换剂可用甲醇或丙酮做项替液。 (⑤开动泵,打开放空阀:待顶替液从放空阀出口流出即关闭阀,压力保持在300一400 200kg/cm2时,说明匀浆液己被顶替液置换 、柱子转 在该压力下保持一段时间 (6)将泵流量调至最小,停泵,卸下柱子即可。 (六)检 检测器是用于连续检测柱流出物中不同组分及其含量的部件:在农药分析中用得最多的 是紫外检测器,相当于把紫外分光光度计连接到色谱柱后。对于在特定波长下有较大吸收峰 的样品 有很高的灵敏度 它不易受温度和流速波动的影 ,有低噪声的特性 ,早期使用 波长紫外检测器的工作波长为254m,以低压汞灯为光源,结构荷单、稳定性好,但对在 254m附近没有吸收的化合物不敏感,适用范围小。现在使用可调波长的紫外可见分光检测 器,工作波长可在190一700m范围内任选,应用范用大大增加,以氘灯和钨灯为光源 关外分光光度法原理及常用溶剂的紫外吸收请参阅本书第六章。此外还有差示折光检测 器,是测量样品和流 相系统 总的折光指数。为了得到 的响应,采用差分技术补偿波 相的折光指数,任何物质只要其折光指数与洗脱液有足够的差别,就可以检测,所以适用范 围广,但灵敏度低,选择性差,受温度干扰较大在农药分析中不常用。 判定检测器性能的指标有以下几个:即灵敏度、噪声和漂移、检测限、线性范围等。① 灵敏度是以组分响应曲线的斜率来表示,即通过检测器的农药数量变化时,相应的响应值变 化率,不同农药的响应曲线斜率是不同的,斜率愈大,表示灵敏度愈高。②噪声是指与农药 无关的输出信号的变化,除了由于仪器的电子系统、电源电压或温度的波动外 洗脱液的 泡和污染可能是出现噪声的主要原因。③漂移是基线随时间增加而产生的定向缓慢变化,噪 声和漂移都直接影响检测能力和分析工作的误差。④色谱峰高度为最大噪声的2倍时,为最 小检测限。⑥线性范围是检测器输出信号与被测组分量呈线性关系的范围。 (七控温系统 早期液相色谱大多采用常温操作 现在随着色谱技术的进展,逐渐重视升温技术 般说米,温度低一些,能提高分离度,但高温可以加快分析速度,因此要综合考虑以上两个 因素,在液固吸附色谱和液液分配色谱中,温度恒定是极为重要的,因为温度的改变,会产 生不均匀吸附和不均匀分配,故液相色谱中不采用程序升温分析,色谱填料往往随温度升降
外,最简单的办法是在分析柱前连接—根 3—5cm 长的保护柱,内装与色谱柱性能相同的填 料,使用 0.2µm 的过滤片,放在进样器和分析柱之间。保护柱可防止来自流动相和样品中 的不溶性微粒对色谱挂的堵塞,还可避免硅胶或键合相的流失,可维护柱效,保护柱并不能 无限止地吸附杂质,使用时应观察检测器讯号,如有基线漂移、怪峰或反压增加等现象,说 明保护柱已被污染,应更换填料。 色谱柱的填充方法:通常有干法和匀浆法两种,填装方法取决于填料微粒的大小,直径 大于 20 µm,可用干法填充;与填充气相色谱柱的方法相似。直径 l0 µm 或 5µm 的微粒, 不能用干法装柱,采用匀浆方法将它们填充到色谱柱中,通常有等比重匀浆法,是选择与硅 胶比重相似的溶剂如四溴乙烷等配成浆状液;由于该溶剂毒性大,硅胶对溴有吸附作用,已 不常用,改用二氧六烷和四氯化碳等,国内亦常用等比重匀浆法,其步骤如下: (1)根据柱体积称取所需之载体于三角瓶中,每 g 裁体加 3ml 二氧六环和 6m1 四氯化碳 匀浆液。 (2)将三角瓶置于超声波清洗器中 10 min,至匀浆液呈半透明状无明显结块为止。 (3)将匀浆液倒入匀浆罐中,化学键合相可在匀浆上部加一些水,旋紧顶盖,打开高压 放空阀。 (4)硅胶柱用脱水的己烷做顶替液,正相键合相用己烷做顶替液,反相键合相或离子交 换剂可用甲醇或丙酮做顶替液。 (5)开动泵,打开放空阀;待顶替液从放空阀出口流出即关闭阀,压力保持在 300 一 400 kg/cm2,当压力下降到 100 一 200 kg/cm2 时,说明匀浆液已被顶替液置换,柱子装填完 毕,在该压力下保持一段时间。 (6)将泵流量调至最小,停泵,卸下柱子即可。 (六)检 测 器 检测器是用于连续检测柱流出物中不同组分及其含量的部件;在农药分析中用得最多的 是紫外检测器,相当于把紫外分光光度计连接到色谱柱后。对于在特定波长下有较大吸收峰 的样品,有很高的灵敏度。它不易受温度和流速波动的影响,有低噪声的特性,早期使用单 波长紫外检测器的工作波长为 254nm,以低压汞灯为光源,结构简单、稳定性好,但对在 254nm 附近没有吸收的化合物不敏感,适用范围小。现在使用可调波长的紫外可见分光检测 器,工作波长可在 190 一 700 nm 范围内任选,应用范围大大增加,以氘灯和钨灯为光源, 有关紫外分光光度法原理及常用溶剂的紫外吸收请参阅本书第六章。此外还有差示折光检测 器,是测量样品和流动相系统总的折光指数。为了得到足够的响应,采用差分技术补偿波动 相的折光指数,任何物质只要其折光指数与洗脱液有足够的差别,就可以检测,所以适用范 围广,但灵敏度低,选择性差,受温度干扰较大在农药分析中不常用。 判定检测器性能的指标有以下几个:即灵敏度、噪声和漂移、检测限、线性范围等。① 灵敏度是以组分响应曲线的斜率来表示,即通过检测器的农药数量变化时,相应的响应值变 化率,不同农药的响应曲线斜率是不同的,斜率愈大,表示灵敏度愈高。②噪声是指与农药 无关的输出信号的变化,除了由于仪器的电子系统、电源电压或温度的波动外,洗脱液的气 泡和污染可能是出现噪声的主要原因。③漂移是基线随时间增加而产生的定向缓慢变化,噪 声和漂移都直接影响检测能力和分析工作的误差。④色谱峰高度为最大噪声的 2 倍时,为最 小检测限。⑥线性范围是检测器输出信号与被测组分量呈线性关系的范围。 (七)控温系统 早期液相色谱大多采用常温操作,现在随着色谱技术的进展,逐渐重视升温技术。一 般说来,温度低一些,能提高分离度,但高温可以加快分析速度,因此要综合考虑以上两个 因素,在液固吸附色谱和液液分配色谱中,温度恒定是极为重要的,因为温度的改变,会产 生不均匀吸附和不均匀分配,故液相色谱中不采用程序升温分析,色谱填料往往随温度升降

产生膨胀和收缩,在某些情况下,温度突然变化会损坏柱床,使用时温度应逐渐升高,实验 结束后,应将柱子慢慢降到室温再停系。 四、分 类 根据固定相对样品分子的保留机理不同主要分为以下4种类型。 1.液固吸附色谱是用高比表面积的颗粒作固定相,根据物质对吸附剂活性表面的吸 其固定相是由在组成上与流动相互不相溶的另 种液体或是借化学 键合到颗粒或载体上,物质的分离是由于不同溶质在两个液相中分配系数不同而达到的。 3.离子交换色谱固定相是具有固定电荷的离子交换树脂,是根据移动相中样本的不 同离子对固定相固定电荷部位的亲和力大小不同而进行分离, 4。凝胶色谱或空间排阻色谱周定相为不同孔径的多孔凝胶,是根据多孔凝胶对不同 大小的分子的排阻效应进行分离。排阻是指大分子不能进入小的凝胶孔中而披排阻在凝胶颗 粒之外,而小分子可进入凝胶内部,在分离过程中,大分子随流动相 “起移动得较 小 子进入凝胶内部移动较慢,这样大小分子可以分开,其分离是严格按照分子尺寸进行的,适 用于非离子型的、分子量高的化合物的分离或分子大小差别很大的样本分离,在农药常量分 析中很少应用。 本章将者重讨论前三类液相色谱方法,上述分离机理,除用于分离分析色谱,还可用于 制备色谱法,制各色谱是制备色谱纯物 的技术 用以分离: 收集 多种色谱纯组 供定性鉴定或作色谱标样,它与分离分析色谱的主要区别是色谱柱尺寸大,柱容量高,可分 离的样品量高几个数量级。制备色谱有两种类型,一类是专用制备色谱仪,色谱系统和技术 与常规色谱有较大差异,制备样品量为0.1一1000g:另一类是在分析色谱仪上进行,色语 柱按比例放大,色谱条件与分析分离大致相同,制备量为0.1一25mg )液固吸附色谱法 液固吸附色谱是液相色谱中研究和应用得最早的一种,以具有高比表面积的颗粒作为固 定相。根据样本中各组分对固定相表面吸附作用的差异进行分离和分析的方法,适用于分离 溶于有机溶剂、具有中等分子量、非离子型的化合物,对于具有不同官能团或不同数目的官 能团的组分分子,具右很好的选轻性,还特别话用于异构体的分离 在装填有吸附剂的层析柱内 当流动相流过吸附剂时,吸附剂表面被溶剂分子S)以单 分子层形式完全占有 当样本分子X)(或溶质分子)被流动相带人柱内,在移动的过程中便 扩散到吸附剂表面和微孔内,并在所有表面上与已结合的溶剂分子($)进行竞争吸附,见 图5一5。 X十S时X限用十S (5-12》 虎动根 固定相 溶质分子 图5-5液固吸附色谱模型 如果试样分子(X)对吸附剂表面吸附基团的(如硅胶的硅羟基)吸引力(包括色散力、诱导 力,氢键等)大于溶剂分子(S),溶剂分子被排斥,试样分子被吸附,则X取代S而吸附在吸
产生膨胀和收缩,在某些情况下,温度突然变化会损坏柱床,使用时温度应逐渐升高,实验 结束后,应将柱子慢慢降到室温再停泵。 四、分 类 根据固定相对样品分子的保留机理不同主要分为以下 4 种类型。 1.液固吸附色谱 是用高比表面积的颗粒作固定相,根据物质对吸附剂活性表面的吸 附能力的差异而进行分离。 2.液液分配色谱 其固定相是由在组成上与流动相互不相溶的另一种液体或是借化学 键合到颗粒或载体上,物质的分离是由于不同溶质在两个液相中分配系数不同而达到的。 3.离子交换色谱 固定相是具有固定电荷的离子交换树脂,是根据移动相中样本的不 同离子对固定相固定电荷部位的亲和力大小不同而进行分离, 4.凝胶色谱或空间排阻色谱 固定相为不同孔径的多孔凝胶,是根据多孔凝胶对不同 大小的分子的排阻效应进行分离。排阻是指大分子不能进入小的凝胶孔中而披排阻在凝胶颗 粒之外,而小分子可进入凝胶内部,在分离过程中,大分子随流动相一起移动得较快,小分 子进入凝胶内部移动较慢,这样大小分子可以分开,其分离是严格按照分子尺寸进行的,适 用于非离子型的、分子量高的化合物的分离或分子大小差别很大的样本分离,在农药常量分 析中很少应用。 本章将着重讨论前三类液相色谱方法,上述分离机理,除用于分离分析色谱,还可用于 制备色谱法,制备色谱是制备色谱纯物质的技术。用以分离、收集一种或多种色谱纯组分, 供定性鉴定或作色谱标样,它与分离分析色谱的主要区别是色谱柱尺寸大,柱容量高,可分 离的样品量高几个数量级。制备色谱有两种类型,一类是专用制备色谱仪,色谱系统和技术 与常规色谱有较大差异,制备样品量为 0.1—1000 g;另一类是在分析色谱仪上进行,色谱 柱按比例放大,色谱条件与分析分离大致相同,制备量为 0.1—25mg。 (一)液固吸附色谱法 液固吸附色谱是液相色谱中研究和应用得最早的一种,以具有高比表面积的颗粒作为固 定相。根据样本中各组分对固定相表面吸附作用的差异进行分离和分析的方法,适用于分离 溶于有机溶剂、具有中等分子量、非离子型的化合物,对于具有不同官能团或不同数目的官 能团的组分分子,具有很好的选择性,还特别适用于异构体的分离。 在装填有吸附剂的层析柱内,当流动相流过吸附剂时,吸附剂表面被溶剂分子(S)以单 分子层形式完全占有,当样本分子(X)(或溶质分子)被流动相带人柱内,在移动的过程中便 扩散到吸附剂表面和微孔内,并在所有表面上与已结合的溶剂分子(S)进行竞争吸附,见 图 5-5。 X S X S + 吸附 吸附+ (5-12) 如果试样分子(X)对吸附剂表面吸附基团的(如硅胶的硅羟基)吸引力(包括色散力、诱导 力,氢键等)大于溶剂分子(S),溶剂分子被排斥,试样分子被吸附,则 X 取代 S 而吸附在吸

附剂表面,称为竞争吸附现象。在一定的浓度范围内,这一吸附一一脱吸附过程是热力学平 衡过程(线性等温吸附),这种竞争吸附达到平衡后,可用下式来表示: (5-13) (X)(S级附】 式中K为分配系数。由此式可看出,如果溶剂分子吸附性强,则被吸附的试样分子(X) 相应减少。但是在流动相中流动相(溶剂)分子与样品分子之间亦存在相互作用,这种附加的 作用增加了色谱过程的复杂性,试样分子在吸附剂表面和在滴 相中的存在也达到了动态平 衡,因此试样 谱柱内的保留行为与吸附剂和流动相有关,适当地选择与此 二项有关的 数,可以使试样在柱内获得良好的分离和保留。 1.吸附色谱固定相的类别常用的吸附剂主要有硅胶和氧化铝等,氧化铝有时会发生 催化反应,己很少使用。硅胶吸附剂用得最多。其特点为:①化学性质稳定,不会与试剂发 生反应:②机械性能强,不会溶涨:@由于多孔结构而具有高的线性容量:①在制造过程中 可以控制其物理性能 如比表面 、孔结构等: 吸附色谱主要是利用物质在吸附剂和流动相中的可逆平衡,以及吸附剂对不同物质吸附 力的差异而使物质分离的,因此吸附剂和流动相的选择是吸附色谱成败的关键。同一种吸附 剂如制备方法、处理方法不同,吸附能力也会相应改变,分离情况也不同。改进色谱柱填料 的结构、性能和填充均匀性,可使液相色普状到高速和高效。经典液相色谱使用全多引大颗 粒图5一6,(©,粒度范围广,形状不规则,柱内填充床很难达到均匀和紧密,不均匀铺层 所产生的涡流会引起色增区带明显扩散,此外,这些多孔型吸附剂的深孔显著地廷缓了试样 分子的传质过程,也引起了色谱区带的扩散。 1-24m 探孔 t6, te) O夫表陆收影纯的先图全事玩室大聚驰 为了克服以上缺点,高效液相色谱主要采用以下两种结构型的吸附剂填料图56)。 (1)海壳型硅胶:是在平均粒径为35μm玻璃微球表面上涂覆1一2μm厚的蒲层硅胶,这 就提高了承受高压的机械强度和填充的均匀性:有些薄壳型填料还采用大的孔径,可显著减 低分子在吸附剂深孔中缓慢传质过程所造成的色谱区带扩散,这类填抖称表面多孔珠,多孔 层珠或薄壳珠,薄壳型硅胶的传质过程快,有利于快速分析。其缺点是比表面积小,样本负 荷量小,柱效不够高,它的应用已逐渐减少。 (②)全多孔微粒型硅胶:粒径减小,填料微孔的相对深度也减小,可以加快传质速率, 因此高效液相色谱大多采用全多孔微粒型吸附剂,常用5um和10um二种,筛分范围一般 为士2um左右,最高校效可达800-1000板/m, 它的优点是柱效高,分离能力强, 可使用较短的柱 上样量大 ,可降低最小检测量 2.吸附色谱固定相的性质在液固吸附色谱中控制保留值和选择性的重要因素是吸附 剂的比表面积和表面活性。 (1)比表面积S:吸附作用是发生在相界面上,被吸附物质的量随着吸附表面增大而增加
附剂表面,称为竞争吸附现象。在一定的浓度范围内,这一吸附——脱吸附过程是热力学平 衡过程(线性等温吸附),这种竞争吸附达到平衡后,可用下式来表示: ( )( ) ( )( ) X S K X S 吸附 吸附 = (5-13) 式中 K 为分配系数。由此式可看出,如果溶剂分子吸附性强,则被吸附的试样分子(X) 相应减少。但是在流动相中流动相(溶剂)分子与样品分子之间亦存在相互作用,这种附加的 作用增加了色谱过程的复杂性,试样分子在吸附剂表面和在流动相中的存在也达到了动态平 衡,因此试样在色谱柱内的保留行为与吸附剂和流动相有关,适当地选择与此二项有关的参 数,可以使试样在柱内获得良好的分离和保留。 1.吸附色谱固定相的类别 常用的吸附剂主要有硅胶和氧化铝等,氧化铝有时会发生 催化反应,已很少使用。硅胶吸附剂用得最多。其特点为:①化学性质稳定,不会与试剂发 生反应;②机械性能强,不会溶涨;@由于多孔结构而具有高的线性容量;①在制造过程中 可以控制其物理性能,如比表面、孔结构等。 吸附色谱主要是利用物质在吸附剂和流动相中的可逆平衡,以及吸附剂对不同物质吸附 力的差异而使物质分离的,因此吸附剂和流动相的选择是吸附色谱成败的关键。同一种吸附 剂如制备方法、处理方法不同,吸附能力也会相应改变,分离情况也不同。改进色谱柱填料 的结构、性能和填充均匀性.可使液相色谱达到高速和高效。经典液相色谱使用全多孔大颗 粒图 5—6,(c),粒度范围广,形状不规则,柱内填充床很难达到均匀和紧密,不均匀铺层 所产生的涡流会引起色谱区带明显扩散,此外,这些多孔型吸附剂的深孔显著地廷缓了试样 分子的传质过程,也引起了色谱区带的扩散。 为了克服以上缺点,高效液相色谱主要采用以下两种结构型的吸附剂填料(图 5—6)。 (1)薄壳型硅胶:是在平均粒径为 35µm 玻璃微球表面上涂覆 l—2µm 厚的蒲层硅胶,这 就提高了承受高压的机械强度和填充的均匀性;有些薄壳型填料还采用大的孔径,可显著减 低分子在吸附剂深孔中缓慢传质过程所造成的色谱区带扩散,这类填抖称表面多孔珠,多孔 层珠或薄壳珠,薄壳型硅胶的传质过程快,有利于快速分析。其缺点是比表面积小,样本负 荷量小,柱效不够高,它的应用已逐渐减少。 (2)全多孔微粒型硅胶:粒径减小,填料微孔的相对深度也减小,可以加快传质速率, 因此高效液相色谱大多采用全多孔微粒型吸附剂,常用 5µm 和 10 µm 二种,筛分范围一般 为±2µm 左右,最高校效可达 80000 一 100000 板/m,它的优点是柱效高,分离能力强, 可使用较短的柱子,上样量大,可降低最小检测量。 2.吸附色谱固定相的性质 在液固吸附色谱中控制保留值和选择性的重要因素是吸附 剂的比表面积和表面活性。 (1)比表面积 S:吸附作用是发生在相界面上,被吸附物质的量随着吸附表面增大而增加
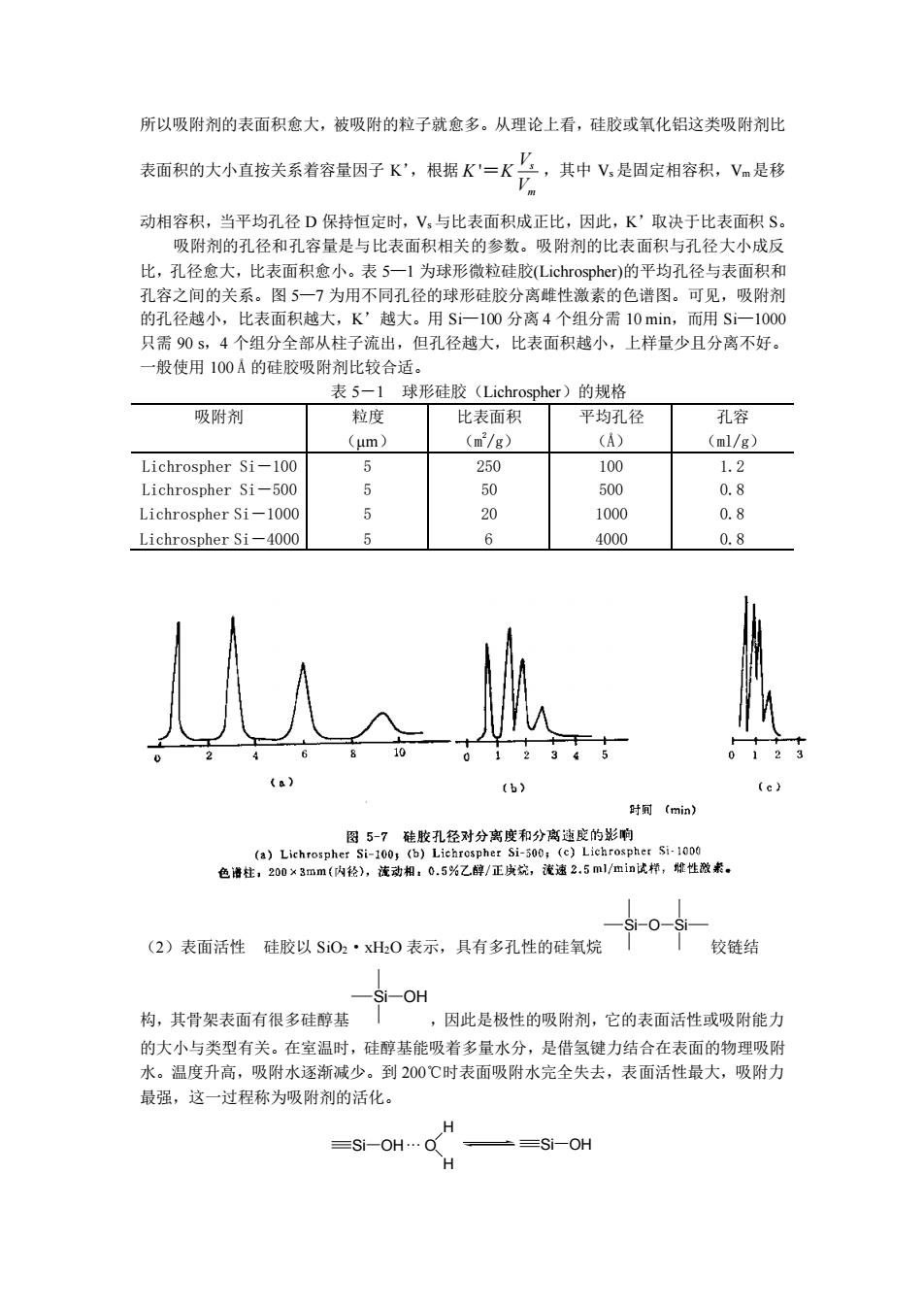
所以吸附剂的表面积愈大,被吸附的粒子就愈多。从理论上看,硅胶或氧化铝这类吸附剂比 表面积的大小直按关系着容量因子K,根据K"=K兰,其中V,是因定相容积,V是移 动相容积,当平均孔径D保持恒定时,V,与比表面积成正比,因此,K'取决于比表面积S。 吸附剂的孔径和孔容量是与比表面积相关的参数。吸附剂的比表面积与孔径大小成反 比,孔径愈大,比表面积愈小。表5一1为球形微粒硅胶Lichrospher)的平均孔径与表面积和 孔容之 的关系。 为用不 同孔径的球形硅胶分离雌性激素的色谱图。可见,吸附济 的孔径越小,比表面积越大,K,越大。用Si一100分离4个组分需10min,而用Si-1000 只需90s,4个组分全部从柱子流出,但孔径越大,比表面积越小,上样量少且分离不好。 一般使用100A的硅胶吸附剂比较合适。 表5一1球形硅胶(Lichrospher)的规格 吸附剂 粒度 比表面利 平均孔径 孔容 (um) (■/g (ml/g) Lichrospher si-100 5 250 100 1.2 Lichrospher si-500 50 500 0.8 Lichrospher Si-1000 20 1000 0.8 Lichrospher Si-4000 4000 0.8 是 (b) 时可(mia) -0-Si (2)表面活性硅胶以S0·x0表示,具有多孔性的硅氧烷 「铰链结 Si-OH 构,其骨架表面有很多硅醇基 ,因此是极性的吸附剂,它的表面活性或吸附能力 的大小与类型有关。在室温时,硅醇基能吸着多量水分,是借氢键力结合在表面的物理吸附 水。温度升高,吸附水逐渐减少。到200℃时表面吸附水完全失去,表面活性最大,吸附力 最强,这一过程称为吸附剂的活化。 =S1-OH. Si-OH H
所以吸附剂的表面积愈大,被吸附的粒子就愈多。从理论上看,硅胶或氧化铝这类吸附剂比 表面积的大小直按关系着容量因子 K’,根据 ' s m V K K V = ,其中 Vs 是固定相容积,Vm是移 动相容积,当平均孔径 D 保持恒定时,Vs 与比表面积成正比,因此,K’取决于比表面积 S。 吸附剂的孔径和孔容量是与比表面积相关的参数。吸附剂的比表面积与孔径大小成反 比,孔径愈大,比表面积愈小。表 5—1 为球形微粒硅胶(Lichrospher)的平均孔径与表面积和 孔容之间的关系。图 5—7 为用不同孔径的球形硅胶分离雌性激素的色谱图。可见,吸附剂 的孔径越小,比表面积越大,K’越大。用 Si—100 分离 4 个组分需 10 min,而用 Si—1000 只需 90 s,4 个组分全部从柱子流出,但孔径越大,比表面积越小,上样量少且分离不好。 一般使用 100 Å 的硅胶吸附剂比较合适。 表 5-1 球形硅胶(Lichrospher)的规格 吸附剂 粒度 (μm) 比表面积 (m 2 /g) 平均孔径 (Å) 孔容 (ml/g) Lichrospher Si-100 Lichrospher Si-500 Lichrospher Si-1000 Lichrospher Si-4000 5 5 5 5 250 50 20 6 100 500 1000 4000 1.2 0.8 0.8 0.8 (2)表面活性 硅胶以 SiO2·xH2O 表示,具有多孔性的硅氧烷 Si O Si 铰链结 构,其骨架表面有很多硅醇基 Si OH ,因此是极性的吸附剂,它的表面活性或吸附能力 的大小与类型有关。在室温时,硅醇基能吸着多量水分,是借氢键力结合在表面的物理吸附 水。温度升高,吸附水逐渐减少。到 200℃时表面吸附水完全失去,表面活性最大,吸附力 最强,这一过程称为吸附剂的活化。 Si OH···O H H Si OH