
In normal atrial,Purkinje,and ventricular cells,the action potential upstroke (phase 0) is dependent on sodium current.From a functional point of view,it is convenient to describe the behavior of the sodium current in terms of three channel states(Figure 2). The cardiac sodium channel protein has been cloned,and it is now recognized that these channel states actually represent different protein conformations.In addition, regions of the protein that confer specific behaviors,such as voltage sensing,pore formation,and inactivation,are now being identified.The gates described below and in Figure 2 represent such regions. Depolarization to the threshold voltage results in opening of the activation (m)gates of sodium channels(Figure 2,middle).If the inactivation (h)gates of these channels have not already closed,the channels are now open or activated,and sodium permeability is markedly increased,greatly exceeding the permeability for any other ion.Extracellular sodium therefore diffuses down its electrochemical gradient into the cell,and the membrane potential very rapidly approaches the sodium equilibrium potential,ENa (about +70 mV when Nae 140 mmol/L and Nai 10 mmol/L).This intense sodium current is very brief because opening of the m gates upon depolarization is promptly followed by closure of the h gates and inactivation of the sodium channels(Figure 2,right). Most calcium channels become activated and inactivated in what appears to be the same way as sodium channels,but in the case of the most common type of cardiac calcium channel (the "L"type),the transitions occur more slowly and at more positive potentials.The action potential plateau(phases 1 and 2)reflects the turning off of most of the sodium current,the waxing and waning of calcium current,and the slow development of a repolarizing potassium current. Final repolarization (phase 3)of the action potential results from completion of sodium and calcium channel inactivation and the growth of potassium permeability, so that the membrane potential once again approaches the potassium equilibrium potential.The major potassium currents involved in phase 3 repolarization include a rapidly activating potassium current(Ik)and a slowly activating potassium current (Iks).These two potassium currents are sometimes discussed together as "Ik."These processes are diagrammed in Figure 3.It is noteworthy that a different potassium current,distinct from Ikr and Iks,may control repolarization in sinoatrial nodal cells. This explains why some drugs that block either Ikr or Iks may prolong repolarization in Purkinje and ventricular cells,but have little effect on sinoatrial nodal repolarization. 6
6 In normal atrial, Purkinje, and ventricular cells, the action potential upstroke (phase 0) is dependent on sodium current. From a functional point of view, it is convenient to describe the behavior of the sodium current in terms of three channel states (Figure 2). The cardiac sodium channel protein has been cloned, and it is now recognized that these channel states actually represent different protein conformations. In addition, regions of the protein that confer specific behaviors, such as voltage sensing, pore formation, and inactivation, are now being identified. The gates described below and in Figure 2 represent such regions. Depolarization to the threshold voltage results in opening of the activation (m) gates of sodium channels (Figure 2, middle). If the inactivation (h) gates of these channels have not already closed, the channels are now open or activated, and sodium permeability is markedly increased, greatly exceeding the permeability for any other ion. Extracellular sodium therefore diffuses down its electrochemical gradient into the cell, and the membrane potential very rapidly approaches the sodium equilibrium potential, ENa (about +70 mV when Nae = 140 mmol/L and Nai = 10 mmol/L). This intense sodium current is very brief because opening of the m gates upon depolarization is promptly followed by closure of the h gates and inactivation of the sodium channels (Figure 2, right). Most calcium channels become activated and inactivated in what appears to be the same way as sodium channels, but in the case of the most common type of cardiac calcium channel (the "L" type), the transitions occur more slowly and at more positive potentials. The action potential plateau (phases 1 and 2) reflects the turning off of most of the sodium current, the waxing and waning of calcium current, and the slow development of a repolarizing potassium current. Final repolarization (phase 3) of the action potential results from completion of sodium and calcium channel inactivation and the growth of potassium permeability, so that the membrane potential once again approaches the potassium equilibrium potential. The major potassium currents involved in phase 3 repolarization include a rapidly activating potassium current (IKr) and a slowly activating potassium current (IKs). These two potassium currents are sometimes discussed together as "IK." These processes are diagrammed in Figure 3. It is noteworthy that a different potassium current, distinct from IKr and IKs, may control repolarization in sinoatrial nodal cells. This explains why some drugs that block either IKr or IKs may prolong repolarization in Purkinje and ventricular cells, but have little effect on sinoatrial nodal repolarization
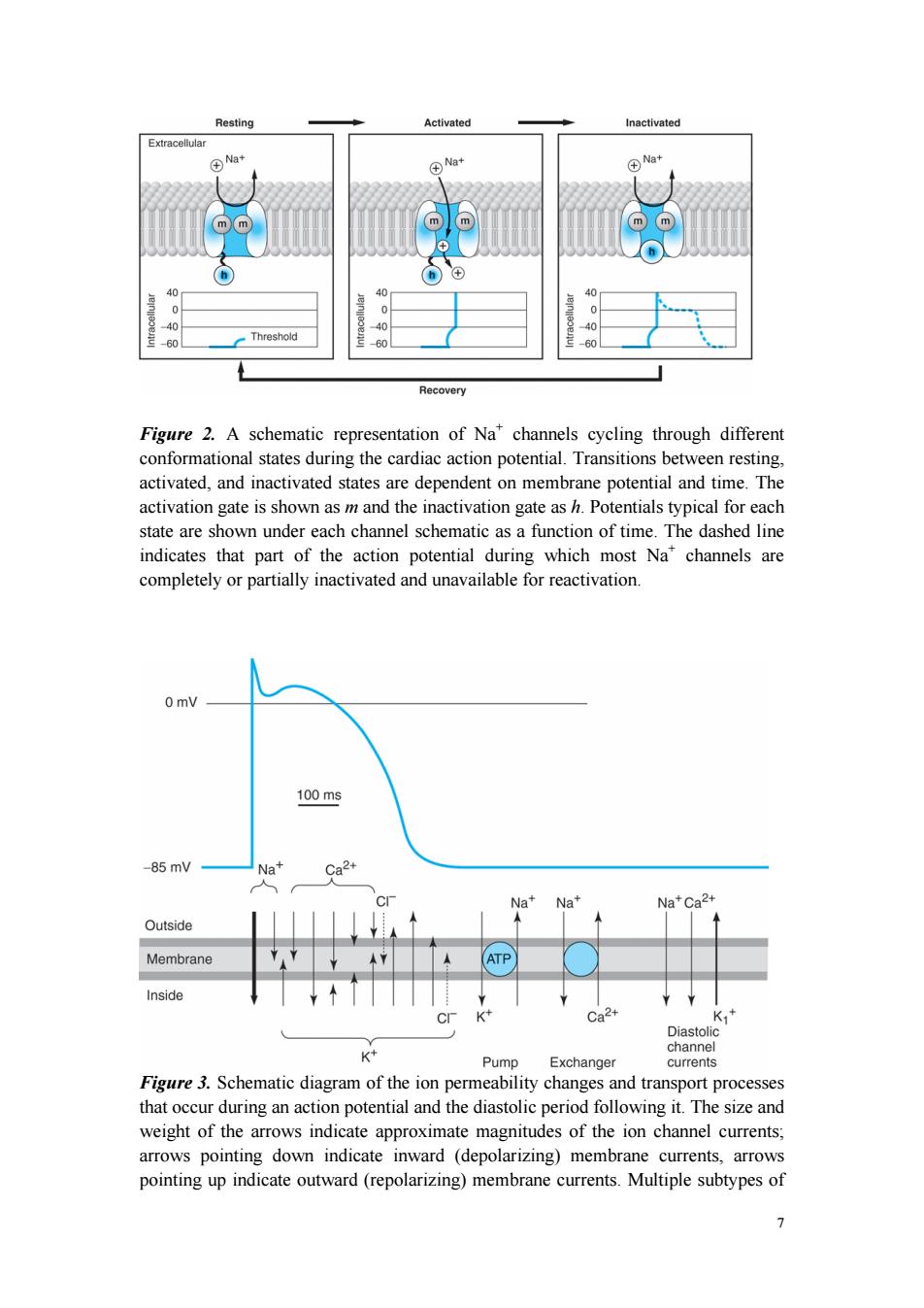
Resting Activated Inactivated Extracellular Na+ ④N+ m m ⑧ 40 0 0 0 90 0 60 Threshold 60 60 Recovery Figure 2.A schematic representation of Na*channels cycling through different conformational states during the cardiac action potential.Transitions between resting, activated,and inactivated states are dependent on membrane potential and time.The activation gate is shown as m and the inactivation gate as h.Potentials typical for each state are shown under each channel schematic as a function of time.The dashed line indicates that part of the action potential during which most Na"channels are completely or partially inactivated and unavailable for reactivation. 0 mV 100ms -85mV Na+ Ca2+ Na+Na+ Na+Ca2+ Outside Membrane ATP Inside Ca2+ K,+ Diastoli K channel Pump Exchanger currents Figure 3.Schematic diagram of the ion permeability changes and transport processes that occur during an action potential and the diastolic period following it.The size and weight of the arrows indicate approximate magnitudes of the ion channel currents; arrows pointing down indicate inward (depolarizing)membrane currents,arrows pointing up indicate outward(repolarizing)membrane currents.Multiple subtypes of 1
7 Figure 2. A schematic representation of Na+ channels cycling through different conformational states during the cardiac action potential. Transitions between resting, activated, and inactivated states are dependent on membrane potential and time. The activation gate is shown as m and the inactivation gate as h. Potentials typical for each state are shown under each channel schematic as a function of time. The dashed line indicates that part of the action potential during which most Na+ channels are completely or partially inactivated and unavailable for reactivation. Figure 3. Schematic diagram of the ion permeability changes and transport processes that occur during an action potential and the diastolic period following it. The size and weight of the arrows indicate approximate magnitudes of the ion channel currents; arrows pointing down indicate inward (depolarizing) membrane currents, arrows pointing up indicate outward (repolarizing) membrane currents. Multiple subtypes of

potassium and calcium currents,with different sensitivities to blocking drugs,have been identified.Chloride currents (dotted arrows)produce both inward and outward membrane currents during the cardiac action potential. The Effect of Resting Potential on Action Potentials A key factor in the pathophysiology of arrhythmias and the actions of antiarrhythmic drugs is the relationship between the resting potential of a cell and the action potentials that can be evoked in it(Figure 4,left panel).Because the inactivation gates of sodium channels in the resting membrane close over the potential range-75 to-55 mV,fewer sodium channels are "available"for diffusion of sodium ions when an action potential is evoked from a resting potential of-60 mV than when it is evoked from a resting potential of-80 mV.Important consequences of the reduction in peak sodium permeability include reduced maximum upstroke velocity (for maximum rate of change of membrane voltage),reduced action potential amplitude,reduced excitability,and reduced conduction velocity. During the plateau of the action potential,most sodium channels are inactivated. Upon repolarization,recovery from inactivation takes place (in the terminology of Figure 2,the h gates reopen),making the channels again available for excitation.The time between phase 0 and sufficient recovery of sodium channels in phase 3 to permit a new propagated response to an external stimulus is the refractory period.Changes in refractoriness (determined by either altered recovery from inactivation or altered action potential duration)can be important in the genesis or suppression of certain arrhythmias.Another important effect of less negative resting potential is prolongation of this recovery time,as shown in Figure 4 (right panel).The prolongation of recovery time is reflected in an increase in the effective refractory period. A brief,sudden,depolarizing stimulus,whether caused by a propagating action potential or by an external electrode arrangement,causes the opening of large numbers of activation gates before a significant number of inactivation gates can close. In contrast,slow reduction (depolarization)of the resting potential,whether brought about by hyperkalemia,sodium pump blockade,or ischemic cell damage,results in depressed sodium currents during the upstrokes of action potentials.Depolarization of the resting potential to levels positive to -55 mV abolishes sodium currents,since all sodium channels are inactivated.However,such severely depolarized cells have been found to support special action potentials under circumstances that increase calcium permeability or decrease potassium permeability.These "slow responses"-slow upstroke velocity and slow conductiondepend on a calcium inward current and constitute the normal electrical activity in the sinoatrial and atrioventricular nodes, since these tissues have a normal resting potential in the range of-50 to-70 mV.Slow responses may also be important for certain arrhythmias.Modern techniques of molecular biology and electrophysiology can identify multiple subtypes of calcium 8
8 potassium and calcium currents, with different sensitivities to blocking drugs, have been identified. Chloride currents (dotted arrows) produce both inward and outward membrane currents during the cardiac action potential. The Effect of Resting Potential on Action Potentials A key factor in the pathophysiology of arrhythmias and the actions of antiarrhythmic drugs is the relationship between the resting potential of a cell and the action potentials that can be evoked in it (Figure 4, left panel). Because the inactivation gates of sodium channels in the resting membrane close over the potential range -75 to -55 mV, fewer sodium channels are "available" for diffusion of sodium ions when an action potential is evoked from a resting potential of -60 mV than when it is evoked from a resting potential of -80 mV. Important consequences of the reduction in peak sodium permeability include reduced maximum upstroke velocity (for maximum rate of change of membrane voltage), reduced action potential amplitude, reduced excitability, and reduced conduction velocity. During the plateau of the action potential, most sodium channels are inactivated. Upon repolarization, recovery from inactivation takes place (in the terminology of Figure 2, the h gates reopen), making the channels again available for excitation. The time between phase 0 and sufficient recovery of sodium channels in phase 3 to permit a new propagated response to an external stimulus is the refractory period. Changes in refractoriness (determined by either altered recovery from inactivation or altered action potential duration) can be important in the genesis or suppression of certain arrhythmias. Another important effect of less negative resting potential is prolongation of this recovery time, as shown in Figure 4 (right panel). The prolongation of recovery time is reflected in an increase in the effective refractory period. A brief, sudden, depolarizing stimulus, whether caused by a propagating action potential or by an external electrode arrangement, causes the opening of large numbers of activation gates before a significant number of inactivation gates can close. In contrast, slow reduction (depolarization) of the resting potential, whether brought about by hyperkalemia, sodium pump blockade, or ischemic cell damage, results in depressed sodium currents during the upstrokes of action potentials. Depolarization of the resting potential to levels positive to -55 mV abolishes sodium currents, since all sodium channels are inactivated. However, such severely depolarized cells have been found to support special action potentials under circumstances that increase calcium permeability or decrease potassium permeability. These "slow responses" - slow upstroke velocity and slow conductiondepend on a calcium inward current and constitute the normal electrical activity in the sinoatrial and atrioventricular nodes, since these tissues have a normal resting potential in the range of -50 to -70 mV. Slow responses may also be important for certain arrhythmias. Modern techniques of molecular biology and electrophysiology can identify multiple subtypes of calcium

and potassium channels.One way in which such subtypes may differ is in sensitivity to drug effects,so drugs targeting specific channel subtypes may be developed in the future. 里10,00 Drug 100 10.000 Control 1000 Drug 100 0 Contro -120-100-80 60 -120-100-80 60 Resting membrane potential(mV) Resting membrane potential(mV) Figure 4.Dependence of sodium channel function on the membrane potential preceding the stimulus.Left:The fraction of sodium channels available for opening in response to a stimulus is determined by the membrane potential immediately preceding the stimulus.The decrease in the fraction available when the resting potential is depolarized in the absence of a drug (control curve)results from the voltage-dependent closure of h gates in the channels.The curve labeled Drug illustrates the effect of a typical local anesthetic antiarrhythmic drug.Most sodium channels are inactivated during the plateau of the action potential.Right:The time constant for recovery from inactivation after repolarization also depends on the resting potential.In the absence of drug,recovery occurs in less than 10 ms at normal resting potentials (-85 to -95 mV).Depolarized cells recover more slowly (note logarithmic scale).In the presence of a sodium channel-blocking drug,the time constant of recovery is increased,but the increase is far greater at depolarized potentials than at more negative ones. MECHANISMS OF ARRHYTHMIAS Introduction Many factors can precipitate or exacerbate arrhythmias:ischemia,hypoxia,acidosis or alkalosis,electrolyte abnormalities,excessive catecholamine exposure,autonomic influences,drug toxicity (eg,digitalis or antiarrhythmic drugs),overstretching of cardiac fibers,and the presence of scarred or otherwise diseased tissue.However,all arrhythmias result from (1)disturbances in impulse formation,(2)disturbances in impulse conduction,or(3)both. 9
9 and potassium channels. One way in which such subtypes may differ is in sensitivity to drug effects, so drugs targeting specific channel subtypes may be developed in the future. Figure 4. Dependence of sodium channel function on the membrane potential preceding the stimulus. Left: The fraction of sodium channels available for opening in response to a stimulus is determined by the membrane potential immediately preceding the stimulus. The decrease in the fraction available when the resting potential is depolarized in the absence of a drug (control curve) results from the voltage-dependent closure of h gates in the channels. The curve labeled Drug illustrates the effect of a typical local anesthetic antiarrhythmic drug. Most sodium channels are inactivated during the plateau of the action potential. Right: The time constant for recovery from inactivation after repolarization also depends on the resting potential. In the absence of drug, recovery occurs in less than 10 ms at normal resting potentials (-85 to -95 mV). Depolarized cells recover more slowly (note logarithmic scale). In the presence of a sodium channel-blocking drug, the time constant of recovery is increased, but the increase is far greater at depolarized potentials than at more negative ones. MECHANISMS OF ARRHYTHMIAS Introduction Many factors can precipitate or exacerbate arrhythmias: ischemia, hypoxia, acidosis or alkalosis, electrolyte abnormalities, excessive catecholamine exposure, autonomic influences, drug toxicity (eg, digitalis or antiarrhythmic drugs), overstretching of cardiac fibers, and the presence of scarred or otherwise diseased tissue. However, all arrhythmias result from (1) disturbances in impulse formation, (2) disturbances in impulse conduction, or (3) both

Disturbances of Impulse Formation The interval between depolarizations of a pacemaker cell is the sum of the duration of the action potential and the duration of the diastolic interval.Shortening of either duration results in an increase in pacemaker rate.The more important of the two, diastolic interval,is determined primarily by the slope of phase 4 depolarization (pacemaker potential).Vagal discharge and -receptor-blocking drugs slow normal pacemaker rate by reducing the phase 4 slope(acetylcholine also makes the maximum diastolic potential more negative).Acceleration of pacemaker discharge is often brought about by increased phase 4 depolarization slope,which can be caused by hypokalemia,-adrenoceptor stimulation,positive chronotropic drugs,fiber stretch, acidosis,and partial depolarization by currents of injury. Latent pacemakers(cells that show slow phase 4 depolarization even under normal conditions,eg,some Purkinje fibers)are particularly prone to acceleration by the above mechanisms.However,all cardiac cells,including normally quiescent atrial and ventricular cells,may show repetitive pacemaker activity when depolarized under appropriate conditions,especially if hypokalemia is also present. Afterdepolarizations (Figure 5)are depolarizations that interrupt phase 3 (early afterdepolarizations,EADs)or phase 4 (delayed afterdepolarizations,DADs). EADs are usually exacerbated at slow heart rates and are thought to contribute to the development of long QT-related arrhythmias.DADs on the other hand,often occur when intracellular calcium is increased.They are exacerbated by fast heart rates and are thought to be responsible for some arrhythmias related to digitalis excess,to catecholamines,and to myocardial ischemia. MOLECULAR GENETIC BASIS OF CARDIAC ARRHYTHMIAS It is now possible to define the molecular basis of several congenital and acquired cardiac arrhythmias.The best example is the polymorphic ventricular tachycardia known as torsade de pointes (shown in Figure 7),which is associated with prolongation of the QT interval (especially at the onset of the tachycardia),syncope, and sudden death.This must represent prolongation of the action potential of at least some ventricular cells (Figure 1).The effect can,in theory,be attributed either to increased inward current (gain of function)or decreased outward current (loss of function)during the plateau of the action potential.In fact,recent molecular genetic studies have identified up to 300 different mutations in at least eight ion channel genes that produce the congenital long QT (LQT)syndrome (Table 1),and each mutation may have different clinical implications.Loss of function mutations in potassium channel genes produce decreases in outward repolarizing current and are responsible for LQT subtypes 1,2,5,6,and 7.HERG and KCNE2 (MiRPI)genes 10
10 Disturbances of Impulse Formation The interval between depolarizations of a pacemaker cell is the sum of the duration of the action potential and the duration of the diastolic interval. Shortening of either duration results in an increase in pacemaker rate. The more important of the two, diastolic interval, is determined primarily by the slope of phase 4 depolarization (pacemaker potential). Vagal discharge and -receptor-blocking drugs slow normal pacemaker rate by reducing the phase 4 slope (acetylcholine also makes the maximum diastolic potential more negative). Acceleration of pacemaker discharge is often brought about by increased phase 4 depolarization slope, which can be caused by hypokalemia, -adrenoceptor stimulation, positive chronotropic drugs, fiber stretch, acidosis, and partial depolarization by currents of injury. Latent pacemakers (cells that show slow phase 4 depolarization even under normal conditions, eg, some Purkinje fibers) are particularly prone to acceleration by the above mechanisms. However, all cardiac cells, including normally quiescent atrial and ventricular cells, may show repetitive pacemaker activity when depolarized under appropriate conditions, especially if hypokalemia is also present. Afterdepolarizations (Figure 5) are depolarizations that interrupt phase 3 (early afterdepolarizations, EADs) or phase 4 (delayed afterdepolarizations, DADs). EADs are usually exacerbated at slow heart rates and are thought to contribute to the development of long QT-related arrhythmias. DADs on the other hand, often occur when intracellular calcium is increased. They are exacerbated by fast heart rates and are thought to be responsible for some arrhythmias related to digitalis excess, to catecholamines, and to myocardial ischemia. MOLECULAR GENETIC BASIS OF CARDIAC ARRHYTHMIAS It is now possible to define the molecular basis of several congenital and acquired cardiac arrhythmias. The best example is the polymorphic ventricular tachycardia known as torsade de pointes (shown in Figure 7), which is associated with prolongation of the QT interval (especially at the onset of the tachycardia), syncope, and sudden death. This must represent prolongation of the action potential of at least some ventricular cells (Figure 1). The effect can, in theory, be attributed either to increased inward current (gain of function) or decreased outward current (loss of function) during the plateau of the action potential. In fact, recent molecular genetic studies have identified up to 300 different mutations in at least eight ion channel genes that produce the congenital long QT (LQT) syndrome (Table 1), and each mutation may have different clinical implications. Loss of function mutations in potassium channel genes produce decreases in outward repolarizing current and are responsible for LQT subtypes 1, 2, 5, 6, and 7. HERG and KCNE2 (MiRP1) genes