
④加入某些金属或其氧化物异丁烯乙烯基醚用HC1引发时不能聚合,只能 发生加成反应。若在反应体系中添加Ni、Co、Fc、Ca或有关的氧化物VOs、PbO 或SO,等即可聚合。这些添加剂可促进HC1电离,且与CI络合使之稳定,这样 就遏止了亲核能力较强的负离子与活性中心的成键反应,而使聚合反应能顺利进 行。 4.2.2.2 Lewis酸 它们都是Friedel-Crafts催化剂,是缺电子的无机化合物,阳离子的主要催化 剂。从工业的角度讲,Lws酸是阳离子聚合反应的最重要的引发剂,可以应用的 Lewis酸包括金属卤化物(如BF3、AICl、AIBr、TiCL4、SnCl4、SbCL、PCls ZnC2)和金属卤氧化物(如POCl,、CrO2Cl、SOCl2、VOCl3)。该类引发剂可以 用于合成高相对分子质量的聚合物 (1)生引发剂一助引发剂体系 使用这类引发剂对要加入共引发剂(或称助尼引发剂)作为质子或碳阳离子的 供给体,L©ws酸能将共引发剂上的未共用电子对通过配位键跃近到它的空轨道上 来,生成加成空物,称为酸一成络合物,或简称络合物。 常见的共引发剂体系举例如下: -H'(BF OH) CHa CH AICL3+H,C-CCI=H,C—C+(AICL,) CHa SnCl,+RCl-±R(snCl5) BF3+(C2Hs)20产C2H5(BF,OC2H与) 关于两种化合物的名称,Kennedy在进一步研究后提出自己的看法,把质子 给体或正离子给体叫做引发剂,而把Lcws酸叫做助引发剂。引发剂和助引发剂 组成一个引发体系,目前这种提法得到了越来越多研究人员的赞同。常见的质子 给体有H2O、HC1、HF、CCl,COOH,常见的正离子给体主要是卤代烃,如(CH3CH2) AICl2、(CH3CH2)2A1C1、A1(CHCH2)3、正氯代丁烷、3-氯-1-丁烯、二苯基氯 甲烷等。 另外,Lwis酸的络合能力很强,能与它们络合的化合物范围很广,如单体、 溶剂等均可与之络合,故使反应复杂化 (2)Lewis酸直接引发 Plesch等人的研究表明,比较强的Lewis酸(如AICl,、AIBr3和TiCl,)可以 单独使用,引发阳离子聚合。按提出的引发机理大致可以分成三种: ①双分子离子化过程: 2AIBr3÷AIBr2(AIBr)

AIBr2*(AIBr2)+M ANBr,M(AIBr) ②单电子离子化过程: TiC4+M→TiCl,M'cI ③烯丙基自行引发机理 Kennedy仔细地研究了阳离子聚合的各种单体的结构,发现含有烯丙基型氢 原子的单体,如异丁烯、异戊二烯、ā.甲基苯乙烯、环戊二烯及茚等化合物,即 使在“极度”干燥下仍能聚合:而不含有烯丙基氢原子的苯乙烯、丁二烯则不能 聚合。从而提出了自行引发机理。 C=C- C-H+MeXn c- +MeXn H(Me掀居原子) 用实验证明自离子化过程是很困难的,因为体系中少量的质子或正离子给体 就会对反应有很大影响,如浓度为103mol/L的水足以使TiCL4和AICl,(在CHC1 中)的引发速率提高10倍。对大部分常规反应体系来说,湿含量常足以导致助引 发作用在引发过程中占压倒的优势。 有文献报道,用立体阻碍大的碱,如吡啶衍生物,可以判定L©wis酸引发的 机理。这种碱具有与质子反应的特性,阻止质子引发,但却不能影响Lcws酸与 单体间的亲电加成。Kennedy及其同事们研究在这种“质子肼”2,6-二叔丁基吡 啶存在下L©wis酸-水的引发反应,结果发现:这种碱明显地降低了单体的转化率, 从几乎100%降至<10%。同时,聚合物的相对分子质量变大,分布变窄。这说明 到目前为止,质子引发反应是主要的形式。 引发剂与共引发剂的不同组合,得到不同引发活性,主要决定于向单体提供 质子的能力。 ④其它引发体系 卤素(常用碘)可以作为活性较大单体的引发剂,如甲氧基苯乙烯,烷基乙 烯基醚,乙烯基咔唑等。反应如下: 2+2 下述组成的阳离子盐也可以作为引发剂: CIO4·BF4 SnCl, (CeHs)C' SbCle C,H,(胰三烯阳离子) 这些阴离子的亲核性很弱,不易核和生长链发生反应,成为无终止反应。 1957年,发现异丁烯在一78C在高能射线(Co0γ射线)作用下,可按阳离

子机理进行聚合,认为射线从单体分子中打出电子,形成单体阳离子自由基: H.C=CICH,y H,G=(CH) 这种引发方法的特点是生长链近旁没有反离子存在,可以研究单独以自由离 子增长的聚合反应机理。 4.2.3链增长反应 链增长反应是生成高聚物的主要基元反应,是一个单体不断插入引发过程所 生成的碳阳离子活性中心和反离子形成的离子对之间的增长过程,即插入增长: CH HC-C+(BF,OH)+H.C=C →H,C-C+…(BFOH) CH, CH, c-cCH CH CH, CHa kp(CH)C-CH-*(BF,OH) kp CH, CH, (CHa)C-CHz-C- (BF:OH) 这个增长反应是否容易进行,取决于碳阳离子的稳定性和烯烃双键的亲核性 (即碱性)。碳阳离子越稳定,烯烃的碱性越强,就越容易加成。阳离子聚合反应 中链增长反应的活化能较低,通常为0.88.0kJ/mol。 单体的结构决定了阳离子聚合的反应性。结构包括取代基的极性和立体阻碍 取代基的推电子作用强,有利于提高双键上电子云的密度,有助于单体亲核结构 的形成,便于进一步与碳阳离子活性中心进行亲电加成。取代基的立体阻碍也影 响着链增长的速率和活性中心的加成方式。在甲苯或二氯甲烷溶剂中,在-79℃, BF3O(CHh引发烷基乙烯基醚聚合,烷基极性对聚合速率的影响顺序为: 叔丁基>异丙基>乙基>正丁基>甲基 此外,反离子对聚合速率的影响能力与它们相应的酸的强度有关。78℃下异 丁烯聚合速率受反离子响,相应的Lewis酸顺序如下 BF3>AlBr3>TiBr4>BBr3>SnCl4 4.2.3.1阳离子聚合中的立构控制 早在1947年,人们己经发现用BF,乙醚络合物在-78℃下引发异丁基乙烯基 醚聚合,得到了结晶性聚合物,后来证明它是全同立构聚合物。并且反应温度降 低,全同结构含量增加:相反,反应温度升高,全同结构含量减少。关于阳离了 聚合中立构规整性聚合物的形成过程有很多种解释,至今未有统一的意见。 4.2.3.2假阳离子聚合 烯烃的阳离子聚合过程中,有时增长活性中心不是碳阳离子,而是共价键结

构,这样的反应称为假阳离子聚合(pseudo cationic polymerization)。最典型的例 子是HC1O:引发苯乙烯在CHC2中的阳离子聚合: -st++CIo -st+CIO, -SIOCIO 自由离子 高羊对 蚧椭健 "CH-CH-O-CIO3 c-a,-o-go, HC=CH 假阳离子聚合与阳离子聚合并不是属于两类截然不同的机理,它们之间没有 根本的区别。不过是不同电离度谱中两端的两类活性中心而已。有些聚合既可以 按假阳离子聚合机理进行,也可以按常见的阳离子聚合方式进行,这完全依赖于 聚合条件。例如:质子酸(如HC1O4)引发或碘引发苯乙烯聚合,当溶剂为二氯 甲烷,聚合温度为-20℃时,聚合过程包括三个连续的过程:第一步为快速、寿命 短的离子聚合:第二步无离子特征:第三步又是离子过程。若温度为-20℃~30℃ 时,无第一步,第三步也很短。另一方面,若温度低至-80℃,则只有第一步。离 子聚合通常得高分子质量的聚合物。而共价键引发聚合得到的聚合物为低分子质 量的齐聚物。用SEC(Size Exclusion Chromatography)可以检测出分子质量和 分布的差别。另有一些假阳离子聚合反应的例子见表4-1。 表4-1假阳离子聚合反应的例了 引发剂 溶剂 HCIO HCIO EINO 养乙烯 CF.COOH H20·snCl CH2Cl2 CeHsCH(CHs)CIO CH2Cl2 对甲氧基苯乙烯 CFCOOH CH2Cl2 苊烯 HCIO4 CH2Cl2 2 N-乙烯基咔味 Haso CHCI 1.1-二-对二甲氧基苯乙桥 CCICOOH C6Ha 1,1-二苯基乙烯 HCISbCla CoHe 4.2.3.3异构化聚合 碳阳离子的活性很大,在链增长过程中活性中心可以发生重排反应,转变成
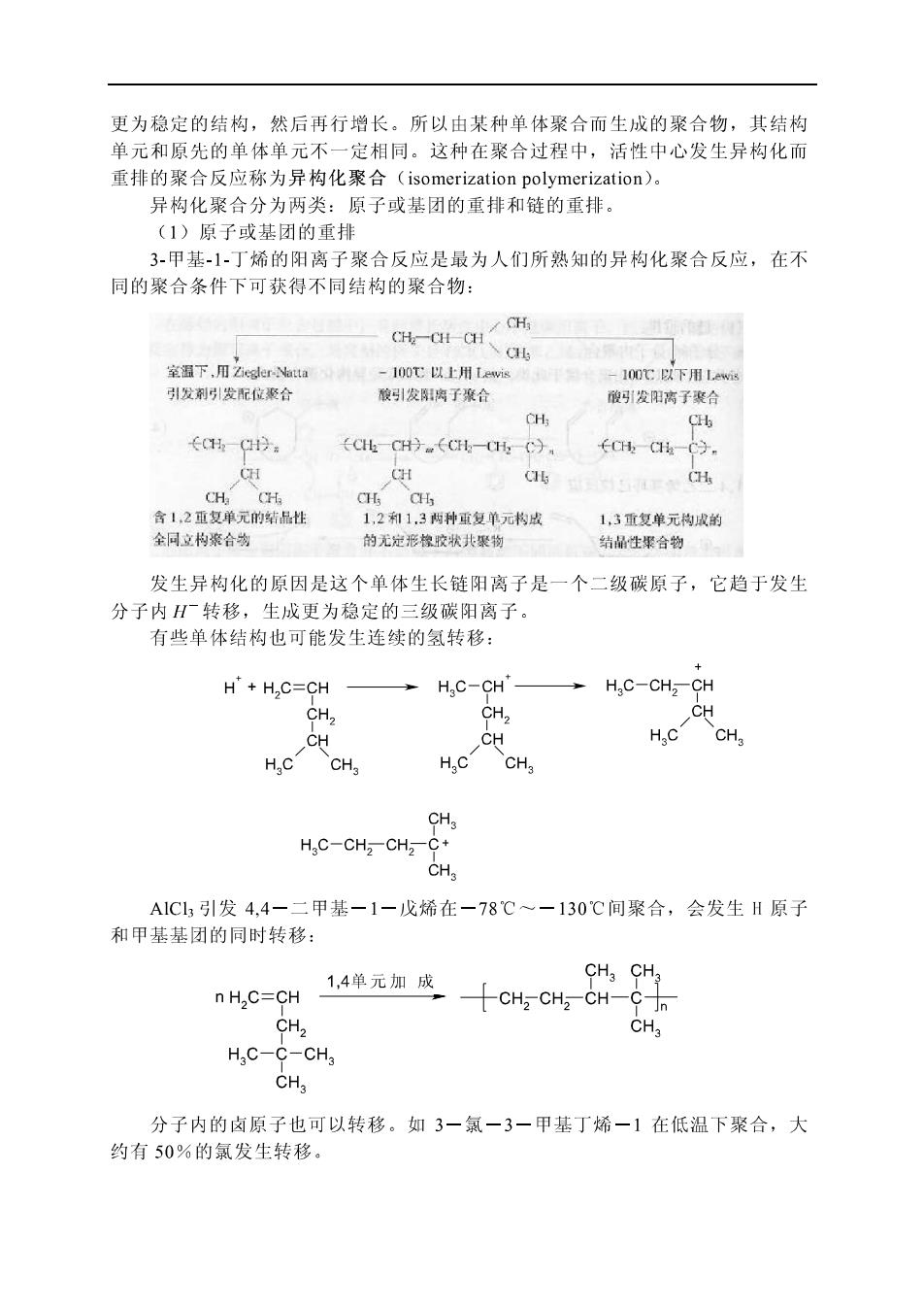
更为稳定的结构,然后再行增长。所以由某种单体聚合而生成的聚合物,其结构 单元和原先的单体单元不一定相同。这种在聚合过程中,活性中心发生异构化而 重排的聚合反应称为异构化聚合(isomerization polymerization)。 异构化聚合分为两类:原子或基团的重排和链的重排。 (】)原子或基团的重非 3甲基-1丁烯的阳离子聚合反应是最为人们所熟知的异构化聚合反应,在不 同的聚合条件下可获得不同结构的聚合物: 室温下乙e 100℃以上用wi H 引发刺引发配位聚合 酸引发阳了合 很雪引发阳高了襄 ←CHC9 (CICH9.(t-,C)。 CH C 全同立构聚合 1,2和1,3两种雪复单元书成 1,3重复单元构成的 的无定形模胶状共聚物 结的生探合物。 发生异构化的原因是这个单体生长链阳离子是一个二级碳原子,它趋于发生 分子内H转移,生成更为稳定的三级碳阳离子。 有些单体结构也可能发生连续的氢转移: H'+H,C=CH H,C-CH' H,C-CH2-CH CH. CH, CH CH CH HCCHa CH, CH H.C-CHz-CHz-C+ A1C1,引发4,4一二甲基-1一戊烯在一78℃一一130C间聚合,会发生H原子 和甲基基团的同时转移: 1,4单元加成 n H2C=CH CH CH2 CH-C CH2 CH3 H C-C-CHa CHa 分子内的卤原子也可以转移。如3一氯一3一甲基丁烯一1在低温下聚合,大 约有50%的氯发生转移