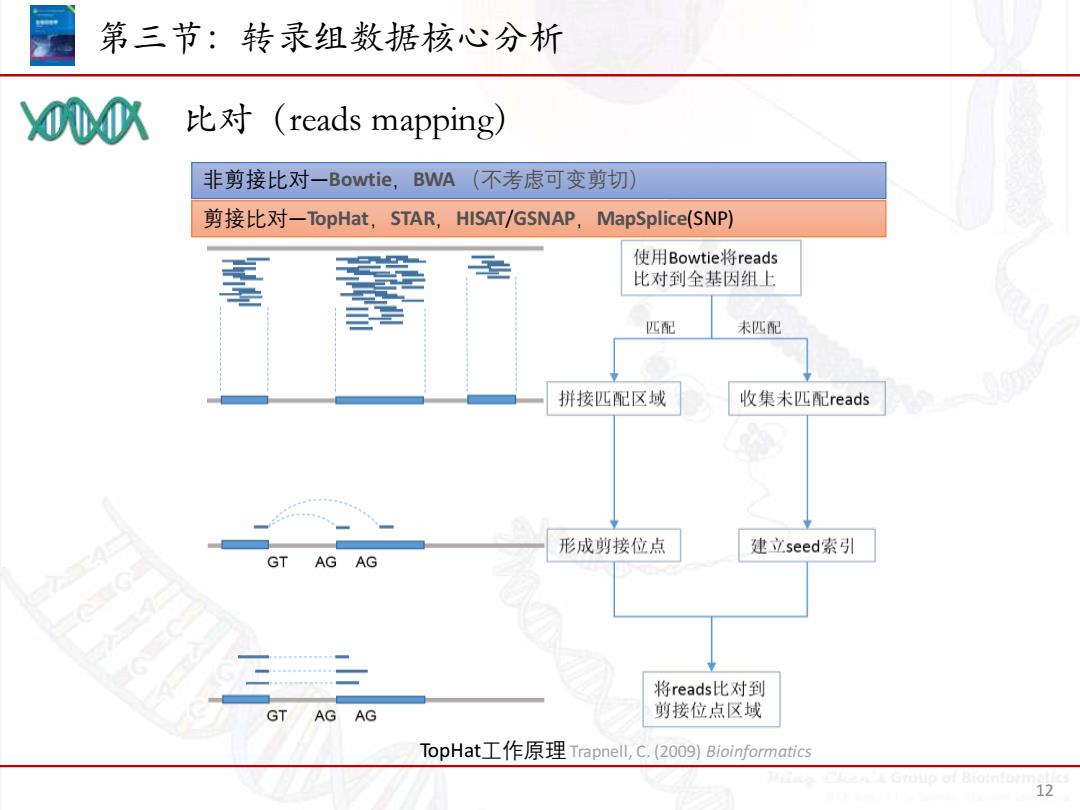
第三节:转录组数据核心分析 000 、比对(reads mapping) 非剪接比对一Bowtie,BWA (不考虑可变剪切) 剪接比对-TopHat,STAR,HISAT/GSNAP,MapSplice(SNP) 使用Bowtie.将reads 比对到全基因组上 匹配 未匹配 拼接匹配区域 收集未匹配reads 形成剪接位点 建立seed索引 GT AGAG 将reads比对到 GT AGAG 剪接位点区域 TopHat.工作原理Trapnell,C,(2009)Bioinformatics 12
第三节:转录组数据核心分析 12 非剪接比对—Bowtie,BWA (不考虑可变剪切) 剪接比对—TopHat,STAR,HISAT/GSNAP,MapSplice(SNP) TopHat工作原理Trapnell, C. (2009) Bioinformatics 比对(reads mapping)
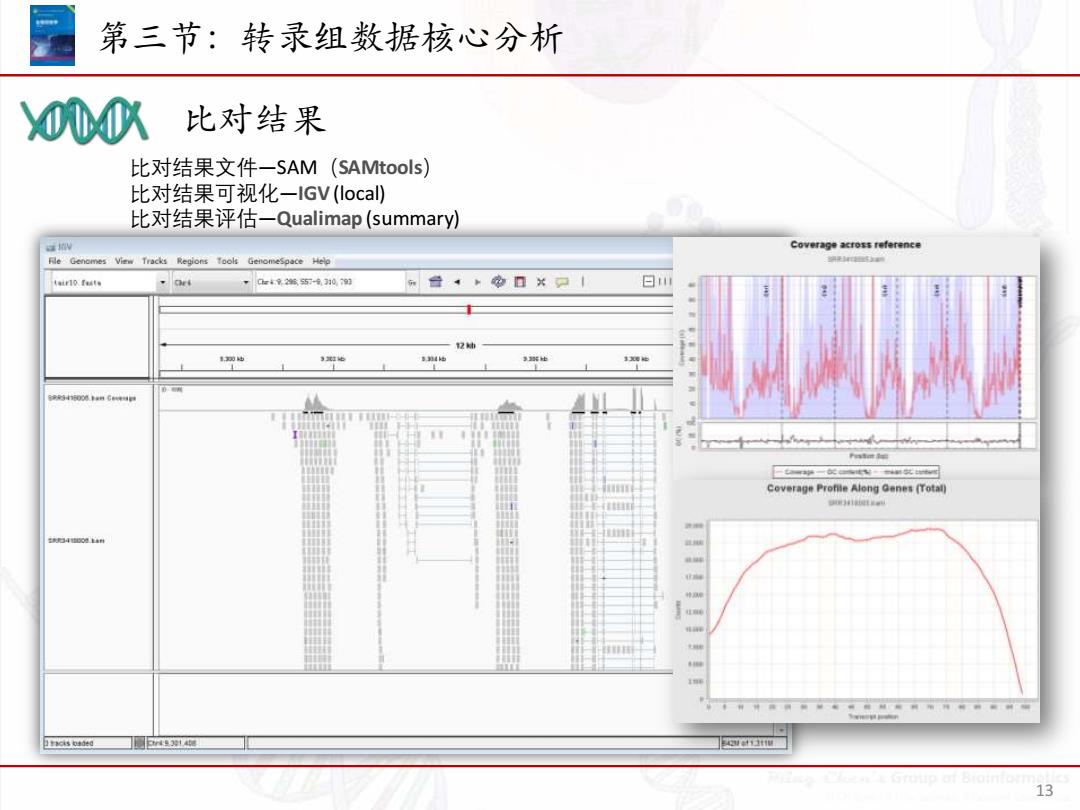
屋 第三节:转录组数据核心分析 000八 比对结果 比对结果文件一SAM(SAMtools) 比对结果可视化一IGV(loca) 比对结果评估一Qualimap(summary) Coverage across reference Fle Genomes View Tracks Regions Tools GenomeSpace Help tair10.Taste 0a402¥s5-43106m 日1 2动 3地 ErageOC ca ttu Coverage Profie Along Genes (Total) 的#1ae 13
第三节:转录组数据核心分析 13 比对结果 比对结果文件—SAM(SAMtools) 比对结果可视化—IGV(local) 比对结果评估—Qualimap (summary)