
第十二章 食品中维生素的测定 【教学目标】: 1.掌握维生素的概念,各种维生素的性质及生理功能和相关知识; 2.了解各类维生素的检测方法,熟练地掌握分光光度计的操作技能。 维生素是人体必需的一类有机营养素。它们的化学组成相互之间差异很大,但对于其 他营养素在体内的正常代谢都起着不可缺少的催化作用。维生素一般不能在体内合成,而 必须从食物中摄取。根据其溶解性,习惯上将维生素分为两大类:一类为脂溶性维生素, 主要包括维生素 A、维生素 D 和维生素 E:一类为水溶性维生素,主要包括 B 族维生素 和维生素 C(即抗坏血酸)。 一、脂溶性维生素的标准测定方法 (一)胡萝卜素的纸层析法(GB12389-90) 本方法适用于植物性食物和含有植物性食物的混合食物中胡萝卜素的测定,其最小检 出限为 0.11μL。 1.原理 以丙酮和石油醚提取食物中的胡萝卜素及其他植物色素;以石油醚为展开剂进行纸层 析,胡萝卜素极性最小,移动速度最快,从而与其他色素分离;剪下含胡萝卜素的区带、 洗脱后于 450nm 波长下定量测定。 2、试剂 (1)石油醚(沸程 30~60℃):同时是展开剂。 (2)丙酮:分析纯。 (3)丙酮+石油醚(3:7)(v/v)。 (4)无水硫酸钠:分析纯。 (5)5% 硫酸钠溶液。 (6)1:1 氢氧化钾溶液:取 50g 氢氧化钾,溶于 50mL 水。 (7)无水乙醇:需经脱醛处理。 (8)β-胡萝卜素标准溶液:取 5mgβ-胡萝卜素标准品,溶于 10ml 三氯甲烷中,浓度约 为 500μg/mL,准确测其浓度。 取标准溶液 10.0μL,加正已烷 3.00ml,混匀,测吸光值,比色杯厚度 1cm,以正 已烷为空白,入射光波长 450nm,平行测定三分,取均值。 计算分式: .(3 21) 0.01 3.01 1000 1 1 = − E A X 式中: X1 -胡萝卜素标准溶液浓度,mg/mL; A-吸光值; E-β-胡萝卜素在正已烷溶液中,入射光波长 450nm,比色杯厚度为 1cm, 溶液浓度为 1μg/ml 的吸光系数,为 0.2638; 1000 1 -将μg/ml 换算成 mg/ml; 0.01 3.01 -测定过程中稀释倍数的换算; (1) β-胡萝卜素标准使用液;将已标定的标准液用石油醚准确稀释后,每毫升溶 液相当 50μg.避光保存于冰箱中。 3.仪器和设备 (1)实验室常用设备。 (2)玻璃层析缸。 (3)分光光度计
第十二章 食品中维生素的测定 【教学目标】: 1.掌握维生素的概念,各种维生素的性质及生理功能和相关知识; 2.了解各类维生素的检测方法,熟练地掌握分光光度计的操作技能。 维生素是人体必需的一类有机营养素。它们的化学组成相互之间差异很大,但对于其 他营养素在体内的正常代谢都起着不可缺少的催化作用。维生素一般不能在体内合成,而 必须从食物中摄取。根据其溶解性,习惯上将维生素分为两大类:一类为脂溶性维生素, 主要包括维生素 A、维生素 D 和维生素 E:一类为水溶性维生素,主要包括 B 族维生素 和维生素 C(即抗坏血酸)。 一、脂溶性维生素的标准测定方法 (一)胡萝卜素的纸层析法(GB12389-90) 本方法适用于植物性食物和含有植物性食物的混合食物中胡萝卜素的测定,其最小检 出限为 0.11μL。 1.原理 以丙酮和石油醚提取食物中的胡萝卜素及其他植物色素;以石油醚为展开剂进行纸层 析,胡萝卜素极性最小,移动速度最快,从而与其他色素分离;剪下含胡萝卜素的区带、 洗脱后于 450nm 波长下定量测定。 2、试剂 (1)石油醚(沸程 30~60℃):同时是展开剂。 (2)丙酮:分析纯。 (3)丙酮+石油醚(3:7)(v/v)。 (4)无水硫酸钠:分析纯。 (5)5% 硫酸钠溶液。 (6)1:1 氢氧化钾溶液:取 50g 氢氧化钾,溶于 50mL 水。 (7)无水乙醇:需经脱醛处理。 (8)β-胡萝卜素标准溶液:取 5mgβ-胡萝卜素标准品,溶于 10ml 三氯甲烷中,浓度约 为 500μg/mL,准确测其浓度。 取标准溶液 10.0μL,加正已烷 3.00ml,混匀,测吸光值,比色杯厚度 1cm,以正 已烷为空白,入射光波长 450nm,平行测定三分,取均值。 计算分式: .(3 21) 0.01 3.01 1000 1 1 = − E A X 式中: X1 -胡萝卜素标准溶液浓度,mg/mL; A-吸光值; E-β-胡萝卜素在正已烷溶液中,入射光波长 450nm,比色杯厚度为 1cm, 溶液浓度为 1μg/ml 的吸光系数,为 0.2638; 1000 1 -将μg/ml 换算成 mg/ml; 0.01 3.01 -测定过程中稀释倍数的换算; (1) β-胡萝卜素标准使用液;将已标定的标准液用石油醚准确稀释后,每毫升溶 液相当 50μg.避光保存于冰箱中。 3.仪器和设备 (1)实验室常用设备。 (2)玻璃层析缸。 (3)分光光度计

(4)旋转蒸发器,具 150mL 球形瓶。 (5)恒温水浴锅。 (6)皂化回馏装置。 (7)点样器或微量注射器。 (8)新华滤纸:定性,快速或中速,101 号。 4.样品的采集和处理 (1)粮食:样品用水洗三次,置 60℃烤箱中烤干,磨粉,贮于塑料瓶内,放一小包樟脑 精,盖紧瓶塞保存,备用。 (2)蔬菜与其他植物性食物:取可食部用水冲洗三次后,用纱布吸去水滴,切碎,用匀浆 器制成匀浆,贮于塑料瓶内,冰箱内保存备用。 5.测定步骤(以下步骤需在避光条件下进行) (1)样品提取 ①取适量样品,相当于原样 1-5g(含胡萝卜素约 20-80μg)的匀浆,粮食样品视其胡萝 卜素含量而定,置 100mL,带塞锥形瓶中,加入丙酮 20mL,石油醚 5mL,振摇 1min,静置 5min,将提取液转入盛有 100mL5% 硫酸钠溶液的分液漏斗中,再与锥形瓶中,加入 10mL, 丙酮+石油醚混合液,振摇 1min,静置 5min,将提取液并入分液漏斗中。如此提取 2-3 次,直至提取液无色为止。 ②植物油和高脂肪样品,需先皂化,取适量样品(小于 10g),加脱醛乙醇 30mL 再加 10mL1: 1,氢氧化钾溶液,回流加热 30min,然后用冰水使之迅速冷却,皂化后样品用石油醚提取, 直至提取液无色为止。 (2)洗涤 ①将提取液[5.(1)①] 静置分层,弃去下层水溶液,反复用 5% 硫酸钠溶液振摇洗涤, 每次约 15mL,直至下层水溶液清亮为止。 ②将皂化后样品提取液[5.(1)②]用水洗涤至中性。 ③将①或②的石油醚提取液通过盛有 10g 无水硫酸钠的小漏斗,漏入球形瓶,用少量石油 醚分数次洗净分液漏斗和无水硫酸钠层内的色素,洗涤液并入球形瓶内。 (3)浓缩与定容 将上述球形瓶内的提取液于旋转蒸发器上减压蒸发,水浴温度为 60℃,蒸发至约 1ml 时,取下球形瓶,用氮气吹干产即加入 2.00mL 石油醚定容,备层析用。 (4)纸层析 ①点样:在 18cm×30cm 滤纸下端距底边 4cm 处作一基线,在基线取 A、B、C、D 四点(如 图 3-8 所示),吸取 0.100-0.400mL 浓缩液[5.(3)] 在 AB 和 CD 间迅速点样. 图 3-8 ②展开:待纸上所点样液自然挥发干后,将滤纸卷为圆筒状,置于预先用石油醚饱和的层 析缸中,进行上行展开。 ③洗脱:待胡萝卜素与其他色素完全分开后,取出滤纸,自然挥发干石油醚,将位于展开 剂前沿的胡萝卜素层析带剪下,产即放入盛有 5mL 石油醚的具塞试管中,用力振摇,使胡 萝卜素完全溶于溶剂中。 (5)比色测定 用 1cm 比色杯,以石油醚调节零点,于 450nm 波长下测吸光度,以其值从标准曲线上
(4)旋转蒸发器,具 150mL 球形瓶。 (5)恒温水浴锅。 (6)皂化回馏装置。 (7)点样器或微量注射器。 (8)新华滤纸:定性,快速或中速,101 号。 4.样品的采集和处理 (1)粮食:样品用水洗三次,置 60℃烤箱中烤干,磨粉,贮于塑料瓶内,放一小包樟脑 精,盖紧瓶塞保存,备用。 (2)蔬菜与其他植物性食物:取可食部用水冲洗三次后,用纱布吸去水滴,切碎,用匀浆 器制成匀浆,贮于塑料瓶内,冰箱内保存备用。 5.测定步骤(以下步骤需在避光条件下进行) (1)样品提取 ①取适量样品,相当于原样 1-5g(含胡萝卜素约 20-80μg)的匀浆,粮食样品视其胡萝 卜素含量而定,置 100mL,带塞锥形瓶中,加入丙酮 20mL,石油醚 5mL,振摇 1min,静置 5min,将提取液转入盛有 100mL5% 硫酸钠溶液的分液漏斗中,再与锥形瓶中,加入 10mL, 丙酮+石油醚混合液,振摇 1min,静置 5min,将提取液并入分液漏斗中。如此提取 2-3 次,直至提取液无色为止。 ②植物油和高脂肪样品,需先皂化,取适量样品(小于 10g),加脱醛乙醇 30mL 再加 10mL1: 1,氢氧化钾溶液,回流加热 30min,然后用冰水使之迅速冷却,皂化后样品用石油醚提取, 直至提取液无色为止。 (2)洗涤 ①将提取液[5.(1)①] 静置分层,弃去下层水溶液,反复用 5% 硫酸钠溶液振摇洗涤, 每次约 15mL,直至下层水溶液清亮为止。 ②将皂化后样品提取液[5.(1)②]用水洗涤至中性。 ③将①或②的石油醚提取液通过盛有 10g 无水硫酸钠的小漏斗,漏入球形瓶,用少量石油 醚分数次洗净分液漏斗和无水硫酸钠层内的色素,洗涤液并入球形瓶内。 (3)浓缩与定容 将上述球形瓶内的提取液于旋转蒸发器上减压蒸发,水浴温度为 60℃,蒸发至约 1ml 时,取下球形瓶,用氮气吹干产即加入 2.00mL 石油醚定容,备层析用。 (4)纸层析 ①点样:在 18cm×30cm 滤纸下端距底边 4cm 处作一基线,在基线取 A、B、C、D 四点(如 图 3-8 所示),吸取 0.100-0.400mL 浓缩液[5.(3)] 在 AB 和 CD 间迅速点样. 图 3-8 ②展开:待纸上所点样液自然挥发干后,将滤纸卷为圆筒状,置于预先用石油醚饱和的层 析缸中,进行上行展开。 ③洗脱:待胡萝卜素与其他色素完全分开后,取出滤纸,自然挥发干石油醚,将位于展开 剂前沿的胡萝卜素层析带剪下,产即放入盛有 5mL 石油醚的具塞试管中,用力振摇,使胡 萝卜素完全溶于溶剂中。 (5)比色测定 用 1cm 比色杯,以石油醚调节零点,于 450nm 波长下测吸光度,以其值从标准曲线上
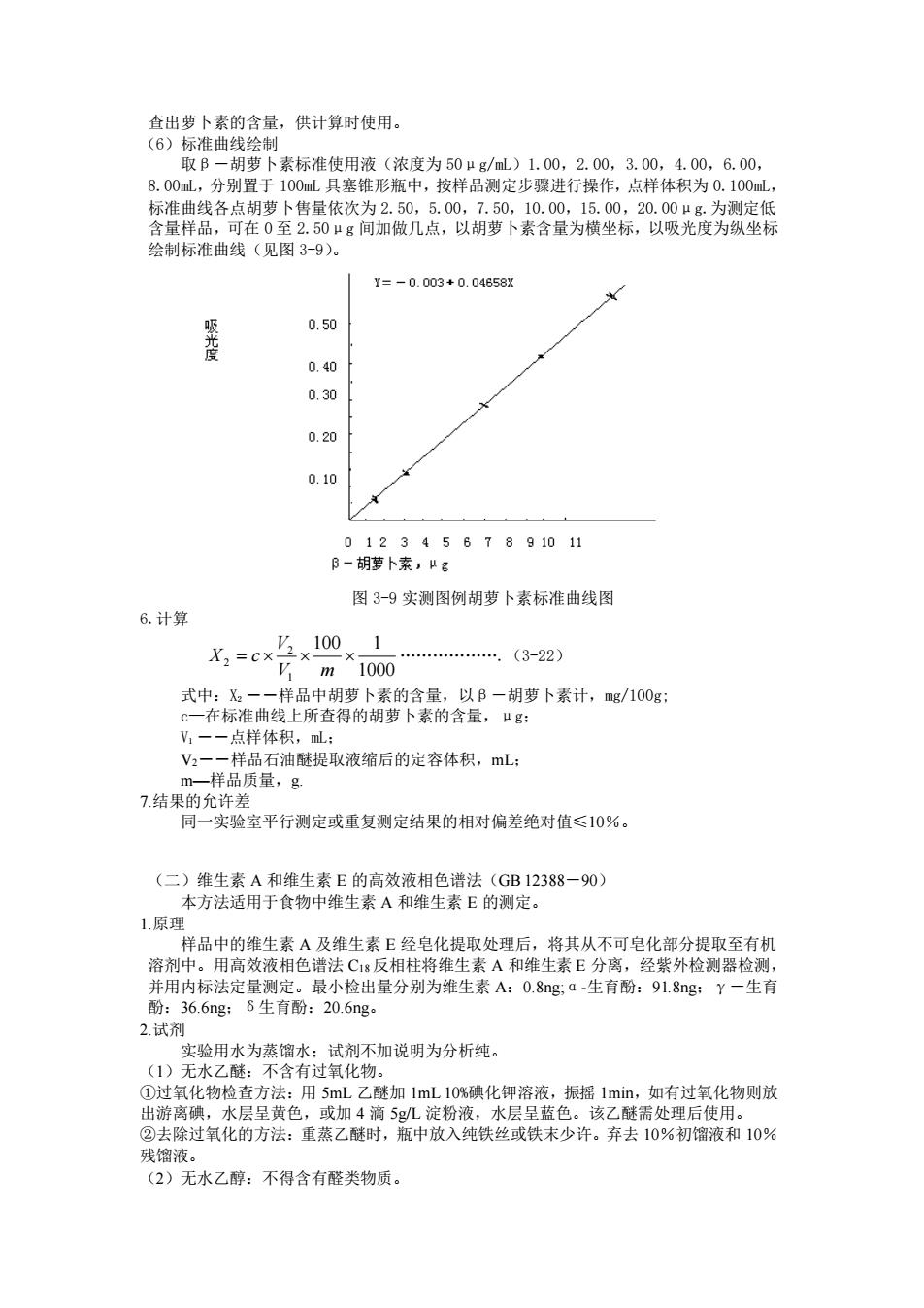
查出萝卜素的含量,供计算时使用。 (6)标准曲线绘制 取β-胡萝卜素标准使用液(浓度为 50μg/mL)1.00,2.00,3.00,4.00,6.00, 8.00mL,分别置于 100mL 具塞锥形瓶中,按样品测定步骤进行操作,点样体积为 0.100mL, 标准曲线各点胡萝卜售量依次为 2.50,5.00,7.50,10.00,15.00,20.00μg.为测定低 含量样品,可在 0 至 2.50μg 间加做几点,以胡萝卜素含量为横坐标,以吸光度为纵坐标 绘制标准曲线(见图 3-9)。 图 3-9 实测图例胡萝卜素标准曲线图 6.计算 1000 100 1 1 2 2 = V m V X c .(3-22) 式中:X2 ――样品中胡萝卜素的含量,以β-胡萝卜素计,mg/100g; c—在标准曲线上所查得的胡萝卜素的含量,μg; V1 ――点样体积,mL; V2――样品石油醚提取液缩后的定容体积,mL; m—样品质量,g. 7.结果的允许差 同一实验室平行测定或重复测定结果的相对偏差绝对值≤10%。 (二)维生素 A 和维生素 E 的高效液相色谱法(GB 12388-90) 本方法适用于食物中维生素 A 和维生素 E 的测定。 1.原理 样品中的维生素 A 及维生素 E 经皂化提取处理后,将其从不可皂化部分提取至有机 溶剂中。用高效液相色谱法 C18 反相柱将维生素 A 和维生素 E 分离,经紫外检测器检测, 并用内标法定量测定。最小检出量分别为维生素 A:0.8ng;α-生育酚:91.8ng;γ-生育 酚:36.6ng;δ生育酚:20.6ng。 2.试剂 实验用水为蒸馏水;试剂不加说明为分析纯。 (1)无水乙醚:不含有过氧化物。 ①过氧化物检查方法:用 5mL 乙醚加 1mL 10%碘化钾溶液,振摇 1min,如有过氧化物则放 出游离碘,水层呈黄色,或加 4 滴 5g/L 淀粉液,水层呈蓝色。该乙醚需处理后使用。 ②去除过氧化的方法:重蒸乙醚时,瓶中放入纯铁丝或铁末少许。弃去 10%初馏液和 10% 残馏液。 (2)无水乙醇:不得含有醛类物质
查出萝卜素的含量,供计算时使用。 (6)标准曲线绘制 取β-胡萝卜素标准使用液(浓度为 50μg/mL)1.00,2.00,3.00,4.00,6.00, 8.00mL,分别置于 100mL 具塞锥形瓶中,按样品测定步骤进行操作,点样体积为 0.100mL, 标准曲线各点胡萝卜售量依次为 2.50,5.00,7.50,10.00,15.00,20.00μg.为测定低 含量样品,可在 0 至 2.50μg 间加做几点,以胡萝卜素含量为横坐标,以吸光度为纵坐标 绘制标准曲线(见图 3-9)。 图 3-9 实测图例胡萝卜素标准曲线图 6.计算 1000 100 1 1 2 2 = V m V X c .(3-22) 式中:X2 ――样品中胡萝卜素的含量,以β-胡萝卜素计,mg/100g; c—在标准曲线上所查得的胡萝卜素的含量,μg; V1 ――点样体积,mL; V2――样品石油醚提取液缩后的定容体积,mL; m—样品质量,g. 7.结果的允许差 同一实验室平行测定或重复测定结果的相对偏差绝对值≤10%。 (二)维生素 A 和维生素 E 的高效液相色谱法(GB 12388-90) 本方法适用于食物中维生素 A 和维生素 E 的测定。 1.原理 样品中的维生素 A 及维生素 E 经皂化提取处理后,将其从不可皂化部分提取至有机 溶剂中。用高效液相色谱法 C18 反相柱将维生素 A 和维生素 E 分离,经紫外检测器检测, 并用内标法定量测定。最小检出量分别为维生素 A:0.8ng;α-生育酚:91.8ng;γ-生育 酚:36.6ng;δ生育酚:20.6ng。 2.试剂 实验用水为蒸馏水;试剂不加说明为分析纯。 (1)无水乙醚:不含有过氧化物。 ①过氧化物检查方法:用 5mL 乙醚加 1mL 10%碘化钾溶液,振摇 1min,如有过氧化物则放 出游离碘,水层呈黄色,或加 4 滴 5g/L 淀粉液,水层呈蓝色。该乙醚需处理后使用。 ②去除过氧化的方法:重蒸乙醚时,瓶中放入纯铁丝或铁末少许。弃去 10%初馏液和 10% 残馏液。 (2)无水乙醇:不得含有醛类物质

①检查方法:取 2mL 银铵溶液于试管中,加入少量乙醇,摇匀,再加入 10%氢氧化钠溶液, 加热。放置冷却后,若有银镜反应则表示乙醇中有醛。 ②脱醛方法:取 2g 硝酸银,溶于少量水中。取 4g 氢氧化钠溶于温乙醇中。将两者倾入 1L 乙醇中,振摇后,放置暗处 2 天(不时摇动,促进反应),经过滤,置蒸馏瓶中蒸馏,弃去 初蒸出的 50mL。当乙醇中含醛较多时,硝酸银用量适当增加。 (3)无水硫酸钠 (4) 甲醇:重蒸后使用。 (5) 重蒸水 水中加少量高猛酸钾,临用前蒸馏。 (6)抗坏血酸溶液(10%)(m/V):临用前进行配制。 (7)1:1 氢氧化钾溶液 氢氧化钠溶液(10%). (8)硝酸银溶液(5%)(m/V) (9)银氨溶液:加氨水至 5%硝酸银溶液中,直至生成的沉淀重新溶解为止,再加 10%氢氧化 钠溶液数滴,如发生沉淀,再加氨水直至溶解。 (10)维生素 A 标准液:视黄醇(纯度 85%)或视黄醇乙酸酯纯度 90%)经皂化处理后使用。 用脱醛乙醇溶解维生素 A 标准品,使其浓度大约为 1mL 相当于 1mg 视黄醇。临用前用紫 外分光光度法分别标定其准确浓度。 (11)维生素 E 标准液:α-生育酚(纯度 95%)γ-生育酚(纯度 95%)δ生育酚(纯度 95%)用脱醛乙醇分别溶解以上三种维生素 E 标准品,使其浓度大约为 1mL 相当于 1mg. 临用前用紫外分光光度法分别标定此三种维生素 E 的准确浓度。 (12)内标溶液:称取苯并[e]芘(纯度 98%),用脱醛乙醇配制成每毫升相当于 10μg 苯并[e] 芘的内标溶液。 (13)pH1-14 试纸 3.仪器和设备 (1)实验室常用设备。 (2)高压液相色谱仪带紫外分光检测器。 (3)旋转蒸发器。 (4)高速离心机。小离心管:具塑料盖 1.5-3.0mL 塑料离心管(与高速离心机配套)。 (5)高纯氮气。 (6)恒温水浴锅。 (7)紫外分光光度计。 4. 操作步骤 (1)样品处理 ①皂化 称取 1-10g 样品(含维生素 A 约 3μg,维生素 E 各异构体约为 40μg)于皂化瓶中, 加 30mL 无水乙醇,进行搅拌,直到颗粒物分散均匀为止。加 5mL100g/L 抗坏血酸,苯 并[e]芘标准液 2.00mL,混匀。加 10mL1:1 氢氧化钾溶液,混匀。于沸水浴上回流 30min 使皂化完全。皂化后立即放入冰水中冷却。 ②提取 将皂化后的样品移入分液漏斗中,用 50mL 水分 2-3 次洗皂化瓶,洗液并入分液漏 斗中。用约 100mL 乙醚分两次洗皂化瓶及其残渣,乙醚液并入分液漏斗中。如有残渣, 可将此液通过有少许脱脂棉的漏斗滤入分液漏斗。轻轻振摇分液漏斗 2min,静置分导, 弃去水层。 ③洗涤 用约 50mL 水洗分液漏斗中的乙醚层,用 pH 试纸检验水层不显碱性(最初水洗轻摇, 逐次振摇强度可增加)。 ④浓缩 将乙醚提取液经过无水硫酸钠(约 5g)滤入与旋转蒸发器配套的 250-300mL 球形 蒸发瓶内,用约 10mL 乙醚冲洗分液漏斗及无水硫酸钠 3 次,并入蒸发瓶内,并将其接至 旋转蒸发器上,于 55℃水浴中减压蒸馏并回收乙醚,待瓶中剩下约 2mL 乙醚时,取下蒸 发瓶,立即用氮气吹掉乙醚。产即加入 2.00mL 乙醇,充分混合,溶解提取物。 ⑤将乙醇液移入一小塑料离心管中[3.(4)],离心 5min(5000r/min).上清液供色谱分析
①检查方法:取 2mL 银铵溶液于试管中,加入少量乙醇,摇匀,再加入 10%氢氧化钠溶液, 加热。放置冷却后,若有银镜反应则表示乙醇中有醛。 ②脱醛方法:取 2g 硝酸银,溶于少量水中。取 4g 氢氧化钠溶于温乙醇中。将两者倾入 1L 乙醇中,振摇后,放置暗处 2 天(不时摇动,促进反应),经过滤,置蒸馏瓶中蒸馏,弃去 初蒸出的 50mL。当乙醇中含醛较多时,硝酸银用量适当增加。 (3)无水硫酸钠 (4) 甲醇:重蒸后使用。 (5) 重蒸水 水中加少量高猛酸钾,临用前蒸馏。 (6)抗坏血酸溶液(10%)(m/V):临用前进行配制。 (7)1:1 氢氧化钾溶液 氢氧化钠溶液(10%). (8)硝酸银溶液(5%)(m/V) (9)银氨溶液:加氨水至 5%硝酸银溶液中,直至生成的沉淀重新溶解为止,再加 10%氢氧化 钠溶液数滴,如发生沉淀,再加氨水直至溶解。 (10)维生素 A 标准液:视黄醇(纯度 85%)或视黄醇乙酸酯纯度 90%)经皂化处理后使用。 用脱醛乙醇溶解维生素 A 标准品,使其浓度大约为 1mL 相当于 1mg 视黄醇。临用前用紫 外分光光度法分别标定其准确浓度。 (11)维生素 E 标准液:α-生育酚(纯度 95%)γ-生育酚(纯度 95%)δ生育酚(纯度 95%)用脱醛乙醇分别溶解以上三种维生素 E 标准品,使其浓度大约为 1mL 相当于 1mg. 临用前用紫外分光光度法分别标定此三种维生素 E 的准确浓度。 (12)内标溶液:称取苯并[e]芘(纯度 98%),用脱醛乙醇配制成每毫升相当于 10μg 苯并[e] 芘的内标溶液。 (13)pH1-14 试纸 3.仪器和设备 (1)实验室常用设备。 (2)高压液相色谱仪带紫外分光检测器。 (3)旋转蒸发器。 (4)高速离心机。小离心管:具塑料盖 1.5-3.0mL 塑料离心管(与高速离心机配套)。 (5)高纯氮气。 (6)恒温水浴锅。 (7)紫外分光光度计。 4. 操作步骤 (1)样品处理 ①皂化 称取 1-10g 样品(含维生素 A 约 3μg,维生素 E 各异构体约为 40μg)于皂化瓶中, 加 30mL 无水乙醇,进行搅拌,直到颗粒物分散均匀为止。加 5mL100g/L 抗坏血酸,苯 并[e]芘标准液 2.00mL,混匀。加 10mL1:1 氢氧化钾溶液,混匀。于沸水浴上回流 30min 使皂化完全。皂化后立即放入冰水中冷却。 ②提取 将皂化后的样品移入分液漏斗中,用 50mL 水分 2-3 次洗皂化瓶,洗液并入分液漏 斗中。用约 100mL 乙醚分两次洗皂化瓶及其残渣,乙醚液并入分液漏斗中。如有残渣, 可将此液通过有少许脱脂棉的漏斗滤入分液漏斗。轻轻振摇分液漏斗 2min,静置分导, 弃去水层。 ③洗涤 用约 50mL 水洗分液漏斗中的乙醚层,用 pH 试纸检验水层不显碱性(最初水洗轻摇, 逐次振摇强度可增加)。 ④浓缩 将乙醚提取液经过无水硫酸钠(约 5g)滤入与旋转蒸发器配套的 250-300mL 球形 蒸发瓶内,用约 10mL 乙醚冲洗分液漏斗及无水硫酸钠 3 次,并入蒸发瓶内,并将其接至 旋转蒸发器上,于 55℃水浴中减压蒸馏并回收乙醚,待瓶中剩下约 2mL 乙醚时,取下蒸 发瓶,立即用氮气吹掉乙醚。产即加入 2.00mL 乙醇,充分混合,溶解提取物。 ⑤将乙醇液移入一小塑料离心管中[3.(4)],离心 5min(5000r/min).上清液供色谱分析

如果样品中维生素含量过少,可用氮气将乙醇液吹干后,再用乙醇重新定容。并记下体积 比。 (2)标准曲线的制备 ①维生素 A 和维生素 E 标准浓度的标定方法 取维生素 A 和各维生素 E 标准液若干微升,分别稀释至 3.00mL 乙醇中,并分别按给 定波长测定各维生素的吸光值。用比吸光系数计算出该维生素的浓度。测定条件如表 3-2 所示。 表 3-2 标准 加入标准的量 S,uL 比吸光系数 E 1% cm 波长 λ,nm 视黄醇 γ-生育酚 δ-生育酚 α-生育酚 10.00 100.0 100.0 100.0 1835 71 92.8 91.2 325 294 298 298 浓度计算: .(3 23) 10 3.00 100 1 3 − = − − E S A X 式中: X1 -某维生素浓度,g/mL; − A -维生素的平均紫外吸光值; S-加入标准的量,μL。 E-某种维生素 1%比吸光系数; 3 10 3.00 − S -标准液稀释倍数。 ②标准曲线的制备 本方法采用内标法定量。把一定量的维生素 A、γ-生育酚、α-生育酚、δ-生育 酚及内标苯并〔e〕芘液混合均匀。选择合适灵敏度,使上述物质的各峰高约为满量程 70%, 为高浓度点。高浓度的 2 1 为低浓度点(其内标苯并〔e〕芘的浓度值不变),用此二种浓 度的混合标准进行色谱分析,结果见色谱图。维生素标准曲线绘制是以维生素峰面积与内 标物峰面积之比为纵坐标,维生素浓度为横坐标绘制,或计算直线回归方程。如有微处理 机装置,则按仪器说明用二点内标法进行定量。 图 3-10 维生素 A 和维生素 E 色谱图 本方法不能将β-E 和γ-E 分开,故γ-E 峰中包含有β-E 峰
如果样品中维生素含量过少,可用氮气将乙醇液吹干后,再用乙醇重新定容。并记下体积 比。 (2)标准曲线的制备 ①维生素 A 和维生素 E 标准浓度的标定方法 取维生素 A 和各维生素 E 标准液若干微升,分别稀释至 3.00mL 乙醇中,并分别按给 定波长测定各维生素的吸光值。用比吸光系数计算出该维生素的浓度。测定条件如表 3-2 所示。 表 3-2 标准 加入标准的量 S,uL 比吸光系数 E 1% cm 波长 λ,nm 视黄醇 γ-生育酚 δ-生育酚 α-生育酚 10.00 100.0 100.0 100.0 1835 71 92.8 91.2 325 294 298 298 浓度计算: .(3 23) 10 3.00 100 1 3 − = − − E S A X 式中: X1 -某维生素浓度,g/mL; − A -维生素的平均紫外吸光值; S-加入标准的量,μL。 E-某种维生素 1%比吸光系数; 3 10 3.00 − S -标准液稀释倍数。 ②标准曲线的制备 本方法采用内标法定量。把一定量的维生素 A、γ-生育酚、α-生育酚、δ-生育 酚及内标苯并〔e〕芘液混合均匀。选择合适灵敏度,使上述物质的各峰高约为满量程 70%, 为高浓度点。高浓度的 2 1 为低浓度点(其内标苯并〔e〕芘的浓度值不变),用此二种浓 度的混合标准进行色谱分析,结果见色谱图。维生素标准曲线绘制是以维生素峰面积与内 标物峰面积之比为纵坐标,维生素浓度为横坐标绘制,或计算直线回归方程。如有微处理 机装置,则按仪器说明用二点内标法进行定量。 图 3-10 维生素 A 和维生素 E 色谱图 本方法不能将β-E 和γ-E 分开,故γ-E 峰中包含有β-E 峰