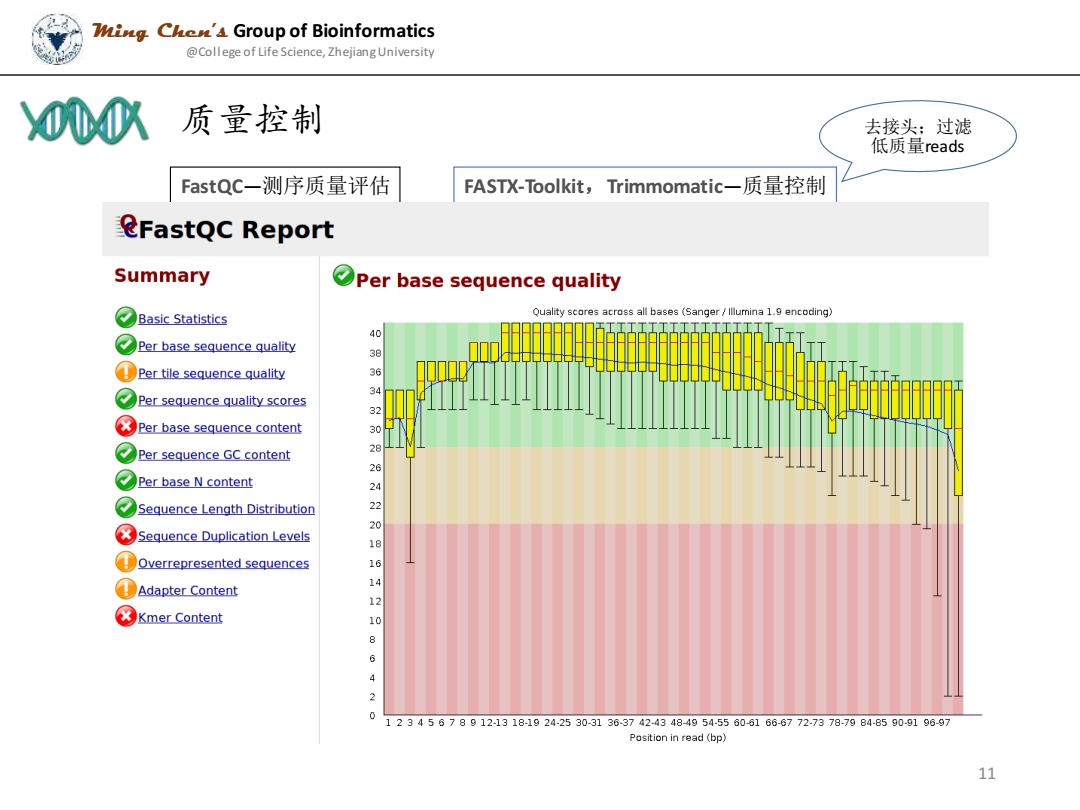
Zig Che'4 Group of Bioinformatics @College of Life Science,Zhejiang University 000 、质量控制 去接头:过滤 低质量reads FastQC--测序质量评估 FASTX-Toolkit,Trimmomatic一质量控制 eFastQC Report Summary Per base sequence quality Basic Statistics Quality scores across all bases (Sanger /lllumina 1.9 encoding) 40 Per base sequence quality Per tile sequence quality 3 Per sequence quality scores Per base sequence content 30 Per sequence GC content ⑦Per base N content 24 Sequence Length Distribution 2220 Sequence Duplication Levels 18 Overrepresented sequences 16 ①Adapter Content 8 Kmer Content 4210 6 4 2 0 12345678912-1318-1924-2530-3136-3742-4348-4954-55606166-6772-7378-79B4-85909196-97 Position in read(bp》 11
FastQC—测序质量评估 FASTX-Toolkit,Trimmomatic—质量控制 质量控制 Ming Chen’s Group of Bioinformatics @College of Life Science, Zhejiang University 11 去接头;过滤 低质量reads
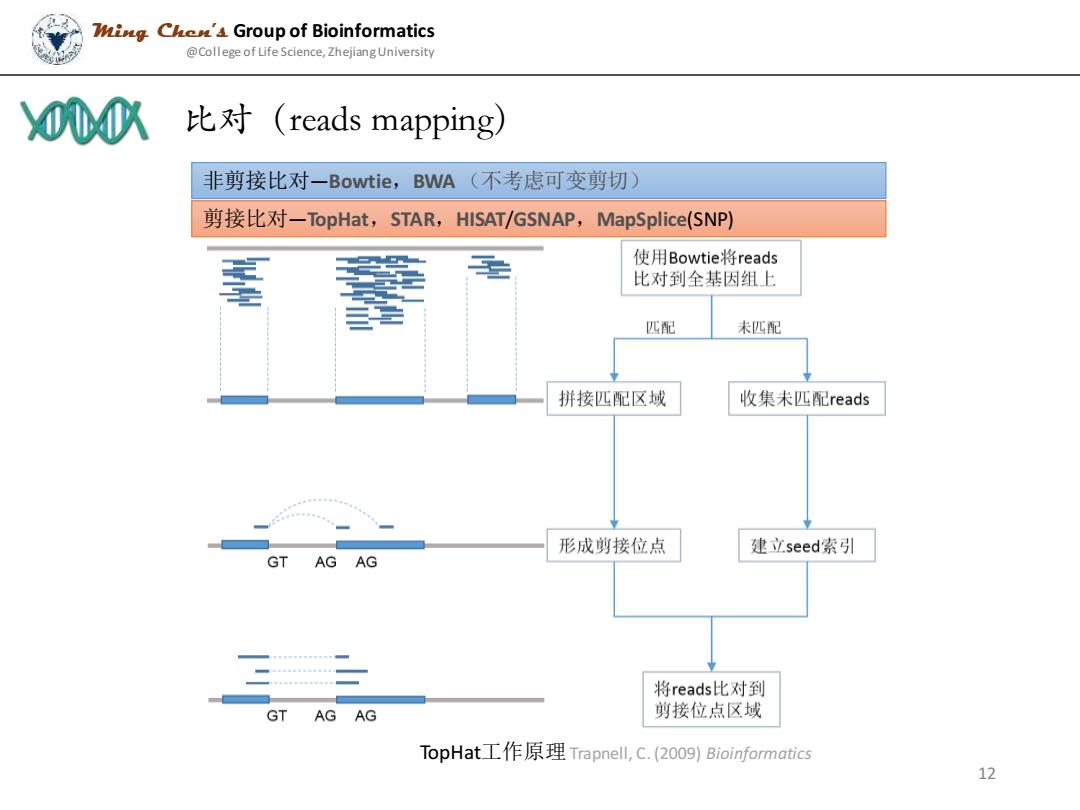
WigChe'4 Group of Bioinformatics @College of Life Science,Zhejiang University 00八 比对(reads mapping) 非剪接比对一Bowtie,BWA(不考虑可变剪切) 剪接比对-TopHat,STAR,HISAT/GSNAP,MapSplice(SNP) 使用Bowtie.将reads 比对到全基因组上 匹配 未匹配 拼接匹配区域 收集未匹配reads 形成剪接位点 建立seed索引 GT AGAG 将reads比对到 GT AG AG 剪接位点区域 TopHat工作原理Trapnell,,C.(20o9)Bioinformatics 12
非剪接比对—Bowtie,BWA (不考虑可变剪切) 剪接比对—TopHat,STAR,HISAT/GSNAP,MapSplice(SNP) TopHat工作原理Trapnell, C. (2009) Bioinformatics 比对(reads mapping) Ming Chen’s Group of Bioinformatics @College of Life Science, Zhejiang University 12