
其它系列填充柱也有类似型号,如Lichrosob RP-18,u-Bondapak C1g等均为键合-C18H7的 填充剂。 加压液相色谱根据所用压力大小不同,可分为高效液相色谱(HPLC,>20个大气压)、中 压液相色谱(MPLC,5~20个大气压)、低压液相色谱(LPLC,<5个大气压)和快速色谱((flash chromatography,约2个大气压)等。其分离效能和分离速度都远高于经典柱色谱。已成为中 药化学成分分离的常规技术手段。 第三节中药有效成分化学结构的研究方法 从中药中经过提取、分离、精制得到的有效成份,必须鉴定或测定其化学结构,才可能 为深入探讨有效成分的生物活性、构效关系、体内代谢以及进行结构改造、人工合成等研究 提供必要的依据。因此,中药有效成分的鉴定和结构测定,是本学科的重要内容之一。 在进行有效成分的结构研究之前,必须对该成分的纯度进行检验,以确证其为单体化学成 分,这是鉴定或测定化学结构的前提。一般常用各种色谱法如薄层色谱(TLC)、纸色谱(PC)、 气相色谱(GC)或高效液相色谱(HPLC)等方法对其进行纯度检验。需要注意的是无论采 用何种方法检验,因为仅用一种溶剂系统或色谱条件,其结论常会出现偏差。在用硅胶薄层 色谱法或高效液相色谱时,最好使用正相和反相薄层或色谱柱同时进行检验,这样可以进一 步保证结论的正确性。此外,固体物质还可通过测定其熔点,考察其熔距的大小作为纯度的 参考。液体物质还可通过测定沸点、沸程、折光率及比重等判断其纯度。对已知物来说,无 论是固体还是液体物质,如其比旋度与文献数据相同,则表明其己是或接近纯品。 一般样品用两种以上溶剂系统或色谱条件进行检测,均显示单一的斑点或谱峰,结晶样 品的熔距为0.5~1.0℃,液体样品的沸程在5℃以内,即认为是较纯的单体化学成分,可用于 化合物的鉴定和结构测定。 在进行有效成分的结构鉴定时,由于同科、同属生物常含有相同或类似的化合物,应对 文献中有关其原生物或近缘生物成分的报道进行调查。并且,在进行提取、分离、精制过程 中可获得对该化合物的部分理化性质(如酸碱性、极性、色谱行为及显色反应等)的认识, 常可为判断该化合物的基本骨架或结构类型提供重要的参考依据。在此基础上,综合运用经 典的理化方法和各种波谱法,对单体化学成分进行鉴定或结构测定。 通过一定的依据判断其可能为己知化合物时,在有对照品的情况下,最好用对照品同时 进行熔点、混合熔点、色谱和红外光谱(R)对照。如果样品与对照品的熔点相同,混合熔 点不降低,色谱中的R值相同,R谱相同,则可判定样品与对照品为同一化合物。 若无对照品,则应多做些数据,或制备衍生物与文献数据核对。如果欲鉴定的化合物为 文献未记载的物质时,应测定该化合物及衍生物的各种波谱并进行必要的化学反应以确定其 化学结构。此时如已推测出该化合物的结构类型,则应充分查找有关该结构类型、结构确定 的最新文献。此外,考察它们的生物合成途径也有助于确定其化学结构。值得提及的是近代 各种波谱法,在鉴定或确定中药有效成分的化学结构中,发挥着极为重要的作用。而经典的 化学方法,由于所需的样品量大,花费时间多,工作量大而复杂,故渐有少用的趋势。但是, 这并不意味着可以完全不需要经典的化学方法。正确的方法是充分认识两种方法在化合物的 鉴定或结构测定的优缺点,灵活运用,使它们相互补充、相互印证,以达到快速而准确无误 地鉴定或测定中药有效成分的化学结构之目的。 一、中药有效成分的理化鉴定 1.物理常数的测定 物理常数的测定包括熔点、沸点、比旋度、折光率和比重等的测定。固体纯物质的熔点, 其熔距应在05~1.0℃的范围内,如熔距过大,则可能存在杂质,应进一步精制或另用不同 14
其它系列填充柱也有类似型号,如Lichrosob RP-18,μ-Bondapak C18等均为键合-C18H37的 填充剂。 加压液相色谱根据所用压力大小不同,可分为高效液相色谱(HPLC,>20 个大气压)、中 压液相色谱(MPLC,5~20 个大气压)、低压液相色谱(LPLC,<5 个大气压)和快速色谱(flash chromatography,约 2 个大气压)等。其分离效能和分离速度都远高于经典柱色谱。已成为中 药化学成分分离的常规技术手段。 第三节 中药有效成分化学结构的研究方法 从中药中经过提取、分离、精制得到的有效成份,必须鉴定或测定其化学结构,才可能 为深入探讨有效成分的生物活性、构效关系、体内代谢以及进行结构改造、人工合成等研究 提供必要的依据。因此,中药有效成分的鉴定和结构测定,是本学科的重要内容之一。 在进行有效成分的结构研究之前,必须对该成分的纯度进行检验,以确证其为单体化学成 分,这是鉴定或测定化学结构的前提。一般常用各种色谱法如薄层色谱(TLC)、纸色谱(PC)、 气相色谱(GC)或高效液相色谱(HPLC)等方法对其进行纯度检验。需要注意的是无论采 用何种方法检验,因为仅用一种溶剂系统或色谱条件,其结论常会出现偏差。在用硅胶薄层 色谱法或高效液相色谱时,最好使用正相和反相薄层或色谱柱同时进行检验,这样可以进一 步保证结论的正确性。此外,固体物质还可通过测定其熔点,考察其熔距的大小作为纯度的 参考。液体物质还可通过测定沸点、沸程、折光率及比重等判断其纯度。对已知物来说,无 论是固体还是液体物质,如其比旋度与文献数据相同,则表明其已是或接近纯品。 一般样品用两种以上溶剂系统或色谱条件进行检测,均显示单一的斑点或谱峰,结晶样 品的熔距为 0.5~1.0℃,液体样品的沸程在 5℃以内,即认为是较纯的单体化学成分,可用于 化合物的鉴定和结构测定。 在进行有效成分的结构鉴定时,由于同科、同属生物常含有相同或类似的化合物,应对 文献中有关其原生物或近缘生物成分的报道进行调查。并且,在进行提取、分离、精制过程 中可获得对该化合物的部分理化性质(如酸碱性、极性、色谱行为及显色反应等)的认识, 常可为判断该化合物的基本骨架或结构类型提供重要的参考依据。在此基础上,综合运用经 典的理化方法和各种波谱法,对单体化学成分进行鉴定或结构测定。 通过一定的依据判断其可能为已知化合物时,在有对照品的情况下,最好用对照品同时 进行熔点、混合熔点、色谱和红外光谱(IR)对照。如果样品与对照品的熔点相同,混合熔 点不降低,色谱中的Rf值相同,IR谱相同,则可判定样品与对照品为同一化合物。 若无对照品,则应多做些数据,或制备衍生物与文献数据核对。如果欲鉴定的化合物为 文献未记载的物质时,应测定该化合物及衍生物的各种波谱并进行必要的化学反应以确定其 化学结构。此时如已推测出该化合物的结构类型,则应充分查找有关该结构类型、结构确定 的最新文献。此外,考察它们的生物合成途径也有助于确定其化学结构。值得提及的是近代 各种波谱法,在鉴定或确定中药有效成分的化学结构中,发挥着极为重要的作用。而经典的 化学方法,由于所需的样品量大,花费时间多,工作量大而复杂,故渐有少用的趋势。但是, 这并不意味着可以完全不需要经典的化学方法。正确的方法是充分认识两种方法在化合物的 鉴定或结构测定的优缺点,灵活运用,使它们相互补充、相互印证,以达到快速而准确无误 地鉴定或测定中药有效成分的化学结构之目的。 一、中药有效成分的理化鉴定 1.物理常数的测定 物理常数的测定包括熔点、沸点、比旋度、折光率和比重等的测定。固体纯物质的熔点, 其熔距应在 0.5~1.0℃的范围内,如熔距过大,则可能存在杂质,应进一步精制或另用不同 14

的溶剂进行重结晶,直至熔点恒定为止。液体物质可测定其沸点。液体纯物质应有恒定的沸 点,除高沸点物质外,其沸程不应超过5℃的范围。此外,液体纯物质还应有恒定的折光率 及比重。比旋度也是物质的一种物理常数。中药的有效成分多为光学活性物质,故无论是己 知还是未知物,在鉴定化学结构时皆应测其比旋度。 少数化合物还需测定其旋光谱和圆二色谱(见本章后述部分)。由于化合物的光学活性 与分子内的立体结构有关,故可利用旋光谱或圆二色谱确定中药有效成分的结构、官能团位 置及分子构象等。 2.分子式的确定 目前最常用的是质谱法(Mass Spectrometry,MS)。高分辨质谱法(High Resolution Mass Spectrometry,HR-MS)不仅可给出化合物的精确分子量,还可以直接给出化合物的分子式。 如青蒿素(qinghaosu)的HR-MS谱中,分子离子峰为m/z282.1472,可计算出其分子式为 C2sH2205(计算值,282.1467)。也可通过质谱中出现的同位素峰的强度推定化合物的分子 式。有时化合物的分子离子峰因不稳定,难以用HR-M$测出,为确定一个化合物的分子式, 需要进行元素定性分析,检查含有哪几种元素,并测定各元素在化合物中所占的百分含量, 从而求出化合物的实验式。元素的定性定量分析过去采用经典的化学方法测定,现在多用自 动元素分析仪测定。前者需要样品量大,且操作复杂:后者则具有快速、简便等优点。得到 一个化合物的实验式后,还要进一步用场解吸质谱、快原子轰击质谱或制备衍生物再测定其 质谱等方法测定它的分子量,以求得化合物的分子式。分子量的测定,以往有很多方法,如 混合熔点降低法、衍生物推导法、酸碱测定法等。但这些方法样品用量大,而且准确性差, 故现己基本不用。 3.化合物的结构骨架与官能团的确定 在决定了一个化合物的分子式后,就需要进行分子结构骨架和官能团的确定。一般首先 决定化合物的不饱和度,准确计算出结构中可能含有的双键数或环数。用化学法推定分子结 构骨架主要依靠后面各章中所述的各类中药化学成分的呈色反应,如羟基蒽醌类化合物通过 碱液显色反应(Borntrager反应)检识:黄酮类化合物可用盐酸镁粉反应、四氢硼钠还原反 应等鉴定:强心苷类化合物可利用甾体母核、ā,B-五元不饱和内酯环和ā-去氧糖的各种 呈色反应结果综合考虑加以判断:苷类化合物则可以通过各种水解反应,然后再以各种呈色 反应及色谱对照分别鉴定生成的苷元及糖的种类等等。官能团的确定也可利用样品与某种试 剂发生颜色变化或产生沉淀等进行判断。在用呈色反应进行分子骨架和官能团检识时最好将 未知样品试验、空白试验及典型样品试验平行进行,以资对照。当根据产生沉淀判断结果时, 要注意液体试样量如过多,会使沉淀现象不明显或沉淀溶解,掩蔽阳性结果:样品分子中含 有两种以上官能团时,可能干扰检识反应。因此,根据一种检识反应的结果尚不足以肯定或 否定该官能团的存在,最好作两种以上的试验,以求得正确的判断。用经典化学方法确定分 子骨架或官能团,有时还要利用其它化学反应如降解反应、氧化反应及还原反应等,甚至通 过化学合成加以验证。 二、中药有效成分的波谱测定 目前,波谱分析等近代技术已成为确定中药有效成分化学结构的主要手段,尤其是最近 发展起来的超导核磁共振技术的普及和各种二维核磁共振谱(Two Dimension Nuclear Magnetic Resonance,2D-NMR)及质谱新技术的开发利用,使其进一步具备了灵敏度高、 选择性强、用量少及快速、简便的优点,大大加快了确定化合物结构的速度和提高了准确性。 红外光谱(R)、紫外光谱(UV)、核磁共振光谱(NMR)和质谱(MS)等波谱分析方 法的基本知识已在分析化学课程中作过介绍,这里仅对这些波谱在中药有效成分结构鉴定中 的应用作简要的介绍。 1.IR光谱 15
的溶剂进行重结晶,直至熔点恒定为止。液体物质可测定其沸点。液体纯物质应有恒定的沸 点,除高沸点物质外,其沸程不应超过 5℃的范围。此外,液体纯物质还应有恒定的折光率 及比重。比旋度也是物质的一种物理常数。中药的有效成分多为光学活性物质,故无论是已 知还是未知物,在鉴定化学结构时皆应测其比旋度。 少数化合物还需测定其旋光谱和圆二色谱(见本章后述部分)。由于化合物的光学活性 与分子内的立体结构有关,故可利用旋光谱或圆二色谱确定中药有效成分的结构、官能团位 置及分子构象等。 2.分子式的确定 目前最常用的是质谱法(Mass Spectrometry,MS)。高分辨质谱法(High Resolution Mass Spectrometry,HR-MS)不仅可给出化合物的精确分子量,还可以直接给出化合物的分子式。 如青蒿素(qinghaosu)的HR-MS谱中,分子离子峰为m/z282.1472,可计算出其分子式为 C25H22O5(计算值,282.1467)。也可通过质谱中出现的同位素峰的强度推定化合物的分子 式。有时化合物的分子离子峰因不稳定,难以用HR-MS测出,为确定一个化合物的分子式, 需要进行元素定性分析,检查含有哪几种元素,并测定各元素在化合物中所占的百分含量, 从而求出化合物的实验式。元素的定性定量分析过去采用经典的化学方法测定,现在多用自 动元素分析仪测定。前者需要样品量大,且操作复杂;后者则具有快速、简便等优点。得到 一个化合物的实验式后,还要进一步用场解吸质谱、快原子轰击质谱或制备衍生物再测定其 质谱等方法测定它的分子量,以求得化合物的分子式。分子量的测定,以往有很多方法,如 混合熔点降低法、衍生物推导法、酸碱测定法等。但这些方法样品用量大,而且准确性差, 故现已基本不用。 3.化合物的结构骨架与官能团的确定 在决定了一个化合物的分子式后,就需要进行分子结构骨架和官能团的确定。一般首先 决定化合物的不饱和度,准确计算出结构中可能含有的双键数或环数。用化学法推定分子结 构骨架主要依靠后面各章中所述的各类中药化学成分的呈色反应,如羟基蒽醌类化合物通过 碱液显色反应(Bornträger 反应)检识;黄酮类化合物可用盐酸镁粉反应、四氢硼钠还原反 应等鉴定;强心苷类化合物可利用甾体母核、α,β-五元不饱和内酯环和α-去氧糖的各种 呈色反应结果综合考虑加以判断;苷类化合物则可以通过各种水解反应,然后再以各种呈色 反应及色谱对照分别鉴定生成的苷元及糖的种类等等。官能团的确定也可利用样品与某种试 剂发生颜色变化或产生沉淀等进行判断。在用呈色反应进行分子骨架和官能团检识时最好将 未知样品试验、空白试验及典型样品试验平行进行,以资对照。当根据产生沉淀判断结果时, 要注意液体试样量如过多,会使沉淀现象不明显或沉淀溶解,掩蔽阳性结果;样品分子中含 有两种以上官能团时,可能干扰检识反应。因此,根据一种检识反应的结果尚不足以肯定或 否定该官能团的存在,最好作两种以上的试验,以求得正确的判断。用经典化学方法确定分 子骨架或官能团,有时还要利用其它化学反应如降解反应、氧化反应及还原反应等,甚至通 过化学合成加以验证。 二、中药有效成分的波谱测定 目前,波谱分析等近代技术已成为确定中药有效成分化学结构的主要手段,尤其是最近 发展起来的超导核磁共振技术的普及和各种二维核磁共振谱(Two Dimension Nuclear Magnetic Resonance,2D-NMR)及质谱新技术的开发利用,使其进一步具备了灵敏度高、 选择性强、用量少及快速、简便的优点,大大加快了确定化合物结构的速度和提高了准确性。 红外光谱(IR)、紫外光谱(UV)、核磁共振光谱(NMR)和质谱(MS)等波谱分析方 法的基本知识已在分析化学课程中作过介绍,这里仅对这些波谱在中药有效成分结构鉴定中 的应用作简要的介绍。 1.IR 光谱 15

用红外光谱法测定结构时,化合物用量只需5~10ug,测定范围在波数4000~500cm间, 其中1600cm以上为化合物的特征基团区,1000~500cm为指纹区。如果被测定物是己知 物,只要和已知对照品做一张共红外光谱图,如果二者红外光谱完全一致,则可推测是同一 物质。如无对照品,也可检索有关红外光谱数据图谱文献。如果被测物结构基本己知,可能 某一局部构型不同,在指纹区就会有差别,如25R与25S型螺甾烷型皂苷元,在960~900cm 附近有显著区别,很容易鉴别。红外光谱对未知结构化合物的鉴定,主要用于功能基的确认, 芳环取代类型的判断等。 2.UV光谱 UV光谱的测定仅需要少量的纯样品,如通常在纸色谱上黄酮类化合物的一个斑点的样 品量,就足够测出几个UV光谱。这对于中药有效成分的研究是非常有利的。 UV光谱在中药有效成分的研究中具有多方面的用途。如与对照品或标准图谱对照,可 用于化合物的初步鉴定:根据Beer-Lambert定律可对中药有效成分进行含量测定,以及根 据中药有效成分的紫外吸收光谱可推定其分子的部分结构等。 一般来说,UV光谱主要可提供分子中的共轭体系的结构信息,可据此判断共轭体系中 取代基的位置、种类和数目。由于UV光谱只能给出分子中部分结构的信息,而不能给出整 个分子的结构信息,所以单独以UV光谱不能决定分子结构,必须与R光谱、NMR谱、 MS谱以及其它理化方法结合才能得到可靠的结论。尽管UV光谱在中药有效成分的结构确 定中提供的信息较少,但对某些具有共轭体系类型的中药有效成分,如蒽醌类、黄酮类以及 强心苷类等成分的结构确定却有重要的实际应用价值。 3.NMR谱 NMR谱是化合物分子在磁场中受电磁波的辐射,有磁距的原子核吸收一定的能量产生 能级的跃迁,即发生核磁共振,以吸收峰的频率对吸收强度作图所得之图谱。它能提供分子 中有关氢及碳原子的类型、数目、互相连接方式、周围化学环境、以及构型、构象的结构信 息。近年随着超导核磁的普及,各种同核(如'HH、3C13C)及异核(如H3C)二维相 关谱的测试与解析技术等的开发应用日新月异,不断得到发展和完善,从而大大加快了结构 测定工作的步伐。目前,分子量在1000以下、几个毫克的微量物质甚至单用NMR测定技术 也可确定它们的分子结构。因此,在进行中药有效成分的结构测定时,NMR谱与其它光谱 相比,其作用最为重要。 (1)lH-NMR谱'H-NMR谱的化学位移(8)范围在O~2Oppm。正常'H-NMR谱技术, 能提供的结构信息参数,主要是化学位移(δ)人、偶合常数)及质子数。'H核因周围化学环境 不同,其外围电子云密度及绕核旋转产生的磁屏蔽效应不同,不同类型的'H核共振信号出现 在不同区域,据此可以识别。偶合常数是磁不等同的两个或两组氢核,在一定距离内因相互 自旋偶合千扰使信号发生裂分,其形状有二重峰(d)、三重峰()、四重峰(q)及多重峰(m)等。 裂分间的距离为偶合常数(。各种不同环境下H核相邻结构具有一定的偶合常数值。除了 正常'H-NMR谱技术外,还有一些帮助结构分析的辅助技术,如选择性去偶、重氢交换、加 入反应试剂、各种双照射等。 应用较多的双照射技术是Nuclear Overhauser Effect(NOE),也称核增益效应。NOE是在 核磁共振中选择地照射一种质子使其饱和,则与该质子在立体空间位置上接近的另一个或数 个质子的信号强度增高的现象。它不但可以找出互相偶合的两个核的关系,还可以反映出不 互相偶合,但空间距离较近的两个核间关系。如五味子酯甲(Schisantherin A)的联苯双酯部分 有二个芳氢,'HNMR示二个单峰,8值分别为6.76、6.43,这两个单峰的归属可用双照射的 NOE技术予以确定。照射83.64的甲氧基,发现位于86.76的芳氢峰增益19%,照射另外三 个甲氧基,均未见到NOE现象,故推测位于甲氧基邻位的芳氢8值为6.76,位于亚甲二氧基 邻位芳氢的8值为6.43. 16
用红外光谱法测定结构时,化合物用量只需5~10μg,测定范围在波数4000~500cm-1间, 其中 1600cm-1以上为化合物的特征基团区,1000~500cm-1为指纹区。如果被测定物是已知 物,只要和已知对照品做一张共红外光谱图,如果二者红外光谱完全一致,则可推测是同一 物质。如无对照品,也可检索有关红外光谱数据图谱文献。如果被测物结构基本已知,可能 某一局部构型不同,在指纹区就会有差别,如 25R与 25S型螺甾烷型皂苷元,在 960~900cm-1 附近有显著区别,很容易鉴别。红外光谱对未知结构化合物的鉴定,主要用于功能基的确认, 芳环取代类型的判断等。 2.UV 光谱 UV 光谱的测定仅需要少量的纯样品,如通常在纸色谱上黄酮类化合物的一个斑点的样 品量,就足够测出几个 UV 光谱。这对于中药有效成分的研究是非常有利的。 UV 光谱在中药有效成分的研究中具有多方面的用途。如与对照品或标准图谱对照,可 用于化合物的初步鉴定;根据 Beer-Lambert 定律可对中药有效成分进行含量测定,以及根 据中药有效成分的紫外吸收光谱可推定其分子的部分结构等。 一般来说,UV 光谱主要可提供分子中的共轭体系的结构信息,可据此判断共轭体系中 取代基的位置、种类和数目。由于 UV 光谱只能给出分子中部分结构的信息,而不能给出整 个分子的结构信息,所以单独以 UV 光谱不能决定分子结构,必须与 IR 光谱、NMR 谱、 MS 谱以及其它理化方法结合才能得到可靠的结论。尽管 UV 光谱在中药有效成分的结构确 定中提供的信息较少,但对某些具有共轭体系类型的中药有效成分,如蒽醌类、黄酮类以及 强心苷类等成分的结构确定却有重要的实际应用价值。 3.NMR 谱 NMR谱是化合物分子在磁场中受电磁波的辐射,有磁距的原子核吸收一定的能量产生 能级的跃迁,即发生核磁共振,以吸收峰的频率对吸收强度作图所得之图谱。它能提供分子 中有关氢及碳原子的类型、数目、互相连接方式、周围化学环境、以及构型、构象的结构信 息。近年随着超导核磁的普及,各种同核(如1 H-1 H、13C-13C)及异核(如1 H-13C)二维相 关谱的测试与解析技术等的开发应用日新月异,不断得到发展和完善,从而大大加快了结构 测定工作的步伐。目前,分子量在 1000 以下、几个毫克的微量物质甚至单用NMR测定技术 也可确定它们的分子结构。因此,在进行中药有效成分的结构测定时,NMR谱与其它光谱 相比,其作用最为重要。 (1)1 H-NMR谱 1 H-NMR谱的化学位移(δ)范围在 0~20ppm。正常1 H-NMR谱技术, 能提供的结构信息参数,主要是化学位移(δ)、偶合常数(J)及质子数。1 H核因周围化学环境 不同,其外围电子云密度及绕核旋转产生的磁屏蔽效应不同,不同类型的1 H核共振信号出现 在不同区域,据此可以识别。偶合常数是磁不等同的两个或两组氢核,在一定距离内因相互 自旋偶合干扰使信号发生裂分,其形状有二重峰(d)、三重峰(t)、四重峰(q)及多重峰(m)等。 裂分间的距离为偶合常数(J)。各种不同环境下1 H核相邻结构具有一定的偶合常数值。除了 正常1 H-NMR谱技术外,还有一些帮助结构分析的辅助技术,如选择性去偶、重氢交换、加 入反应试剂、各种双照射等。 应用较多的双照射技术是Nuclear Overhauser Effect(NOE),也称核增益效应。NOE是在 核磁共振中选择地照射一种质子使其饱和,则与该质子在立体空间位置上接近的另一个或数 个质子的信号强度增高的现象。它不但可以找出互相偶合的两个核的关系,还可以反映出不 互相偶合,但空间距离较近的两个核间关系。如五味子酯甲(Schisantherin A)的联苯双酯部分 有二个芳氢,1 H-NMR示二个单峰,δ值分别为 6.76、6.43,这两个单峰的归属可用双照射的 NOE技术予以确定。照射δ3.64 的甲氧基,发现位于δ6.76 的芳氢峰增益l9%,照射另外三 个甲氧基,均未见到NOE现象,故推测位于甲氧基邻位的芳氢δ值为 6.76,位于亚甲二氧基 邻位芳氢的δ值为 6.43。 16

3.64 OCH3 6.76 H,CO 0 OH H,CO CH3 H,CO …H CH, 6.43 五味子酯甲 (2)13C-NMR谱BC-NMR谱的化学位移范围为0~250ppm,比'H-NMR谱大得多, 是中药化学有效成分结构测定中最重要手段之一。13℃-NMR谱提供的结构信息是分子中各种 不同类型及化学环境的碳核化学位移,异核偶合常数(J)及驰豫时间(T),其中利用度最高 的是化学位移(6c)。常见的3C-NMR测定技术如下。 ①质子宽带去偶:也称质子噪音去偶或全氢去偶。此时H的偶合影响全部被消除,从 而简化了图谱。在分子中没有对称因素和不含F、P等元素时,每个碳原子都会给出一个单 峰,互不重叠。虽无法区别碳上连接H的数,但对判断℃信号的化学位移十分方便。因照射 H后产生NOE现象,连有H的C信号强度增加。季碳信号因不连有H,表现为较弱的峰。 ②偏共振去偶:在偏共振去偶谱中,每个连接质子的碳有残余裂分,故在所得图谱中 次甲基(-CHD碳核呈双峰,亚甲基(-CH2)呈三重峰,甲基(-CH)呈四重峰,季碳为单峰强度 最低。由此可获得碳所连接的质子数、偶合情况等信息。但此法常因各信号的裂分峰相互 重叠,对结构比较复杂的中药有效成分,有些信号难于全部识别或解析,远不及下述的 NEPT和DEPT法易于解析。实际上,后两种方法己基本完全取代了偏共振去偶技术。 ③NEPT(低灵敏核极化转移增强法,insensitive nuclei enhanced by polarization transfer):用调节弛豫时间(△)来调节CH、CH2、CH信号的强度,从而有效地识别CH、 CH2、CH3。季碳因为没有极化转移条件,所以在NEPT谱中无信号。当△=1/4(Jcm)时,CH、 CH2、CH皆为正峰;当△=2/4(JcH)时,只有正的CH峰;当△=34(JcH)时,CH、CH3为 正峰,CH2为负峰。由此可以区分CH、CH和CH信号。再与质子宽带去偶谱对照,还可以确 定季碳信号。 ④DEPT(无畸变极化转移增强法,distortionless enhancement by polarization transfer): 是NEPT的一种改进方法。在DEPT法中,通过改变照射H的脉冲宽度(O),使为45°、90°、 和135°变化并测定C-NMR谱。所得结果与NEPT谱类似。即当0=45时,所有的CH、CH2 CH均显正信号:当0-90°时,仅显示CH正信号:当0=135时,CH和CH为正信号,而CH2为 负信号。季碳同样无信号出现。 图2-10、2-11、2-12分别为B-紫罗兰酮质子宽带去偶谱,偏共振去偶谱及DEPT谱。 17
OCH3 H3CO H3CO O O CH3 OH H CH3 H H3CO O O 6.76 6.43 3.64 五味子酯甲 (2) 13C-NMR谱 13C-NMR谱的化学位移范围为 0~250ppm,比1 H-NMR谱大得多, 是中药化学有效成分结构测定中最重要手段之一。13C-NMR谱提供的结构信息是分子中各种 不同类型及化学环境的碳核化学位移,异核偶合常数(JCH)及驰豫时间(T1),其中利用度最高 的是化学位移(δc)。常见的13C-NMR测定技术如下。 ① 质子宽带去偶: 也称质子噪音去偶或全氢去偶。此时H的偶合影响全部被消除,从 而简化了图谱。在分子中没有对称因素和不含F、P等元素时,每个碳原子都会给出一个单 峰,互不重叠。虽无法区别碳上连接H的数,但对判断13C信号的化学位移十分方便。因照射 H后产生NOE现象,连有H的C信号强度增加。季碳信号因不连有H,表现为较弱的峰。 ② 偏共振去偶: 在偏共振去偶谱中,每个连接质子的碳有残余裂分,故在所得图谱中 次甲基(-CH)碳核呈双峰,亚甲基(-CH2)呈三重峰,甲基(-CH3)呈四重峰,季碳为单峰强度 最低。由此可获得碳所连接的质子数、偶合情况等信息。但此法常因各信号的裂分峰相互 重叠,对结构比较复杂的中药有效成分,有些信号难于全部识别或解析,远不及下述的 INEPT和DEPT法易于解析。实际上,后两种方法已基本完全取代了偏共振去偶技术。 ③ INEPT(低灵敏核极化转移增强法,insensitive nuclei enhanced by polarization transfer):用调节弛豫时间(Δ)来调节CH、CH2、CH3信号的强度,从而有效地识别CH、 CH2、CH3。季碳因为没有极化转移条件,所以在INEPT谱中无信号。当Δ=1/4(JCH)时, CH、 CH2、CH3皆为正峰;当Δ=2/4(JCH)时, 只有正的CH峰;当Δ=3/4(JCH)时, CH、CH3为 正峰, CH2为负峰。由此可以区分CH、CH2和CH3信号。再与质子宽带去偶谱对照,还可以确 定季碳信号。 ④ DEPT(无畸变极化转移增强法,distortionless enhancement by polarization transfer): 是INEPT的一种改进方法。在DEPT法中,通过改变照射1 H的脉冲宽度(θ),使为 45o 、90o 、 和 135o 变化并测定13C-NMR谱。所得结果与INEPT谱类似。即当θ=45o 时,所有的CH、CH2、 CH3均显正信号;当θ=90o 时,仅显示CH正信号;当θ=135o 时,CH和CH3为正信号,而CH2为 负信号。季碳同样无信号出现。 图 2-10、2-11、2-12 分别为 β-紫罗兰酮质子宽带去偶谱,偏共振去偶谱及 DEPT 谱。 17
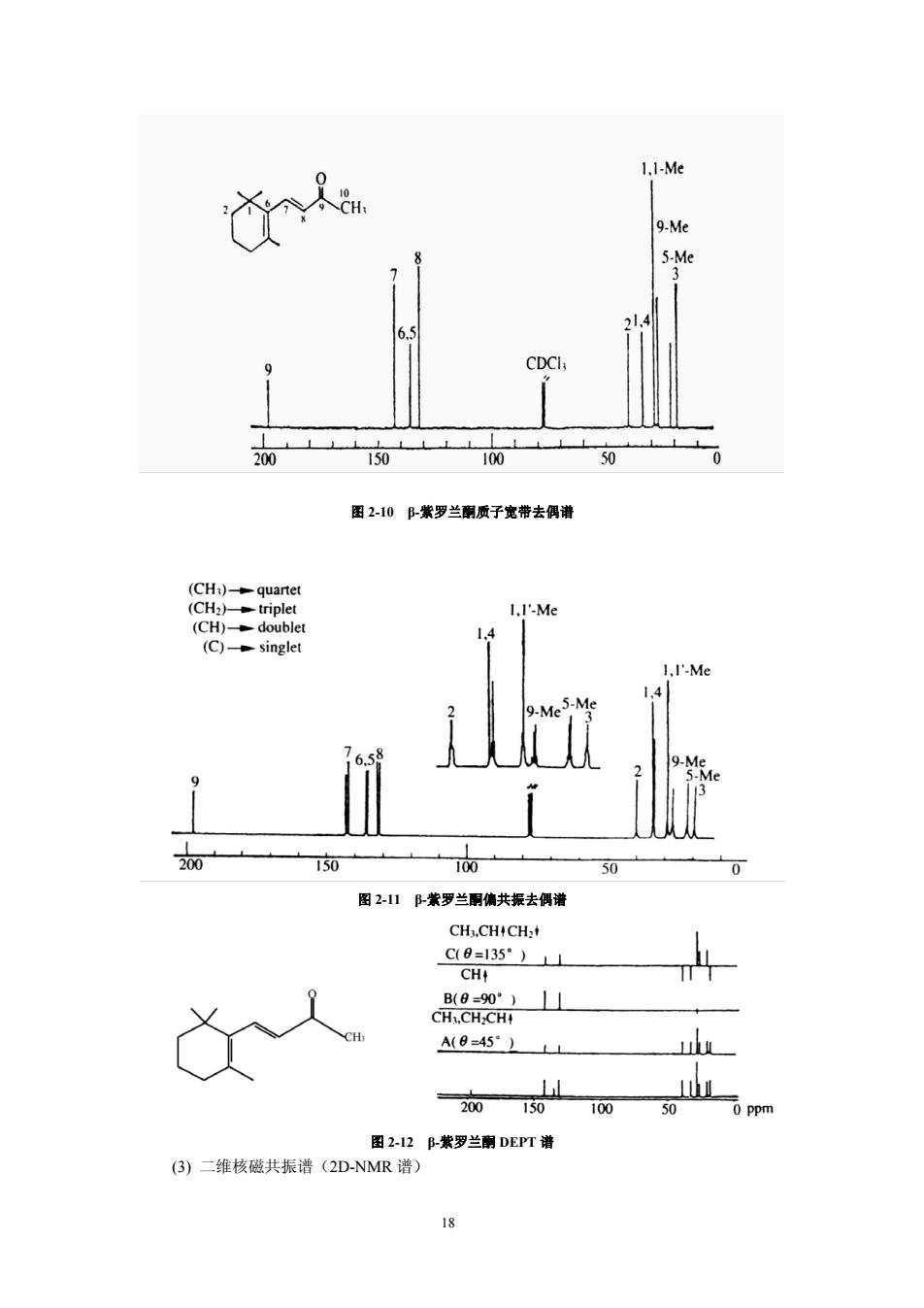
1.1-Me 9-Me 5-Me 3 31.4 6.5 9 CDCI 200 150 100 50 图2-10阝-紫罗兰酮质子宽带去偶谱 (CH3)-quartet (CH2)-triplet 1.1'-Me (CH)-doublet 1.4 (C)-singlet 1,I'-Me 1.4 5-Me 9-Me 7 6.58 9-Me Me 200 150 100 50 0 图2-11B-紫罗兰酮偏共振去偶谱 CH3.CH+CH2+ C(0=135°)1 CH+ B(0-90°)⊥1 CH,CH2CH H A(9=45°) 200 150 100 50 0 Ppm 图2-12B-紫罗兰酮DEPT谱 (3)二维核磁共振谱(2D-NMR谱) 18
图 2-10 β-紫罗兰酮质子宽带去偶谱 图 2-11 β-紫罗兰酮偏共振去偶谱 图 2-12 β-紫罗兰酮 DEPT 谱 (3) 二维核磁共振谱(2D-NMR 谱) 18