
Chapter3.药代动力学PKPD(DMPK) 肝脏 肾脏 第I相反应 (主要P-450) 第1相反应 药物 官能团反应 代谢物 (脂溶性) 结合反应 (水溶性) 代谢物 尿 (水溶性) 胆汁 1991年前40%的受试化合物因为不适宜的药代性质(吸收不完全、代谢太快或 广泛、分布不合理等)而终止
Chapter 3. 药代动力学PKPD( DMPK) 1991年前40%的受试化合物因为不适宜的药代性质(吸收不完全、代谢太快或 广泛、分布不合理等)而终止
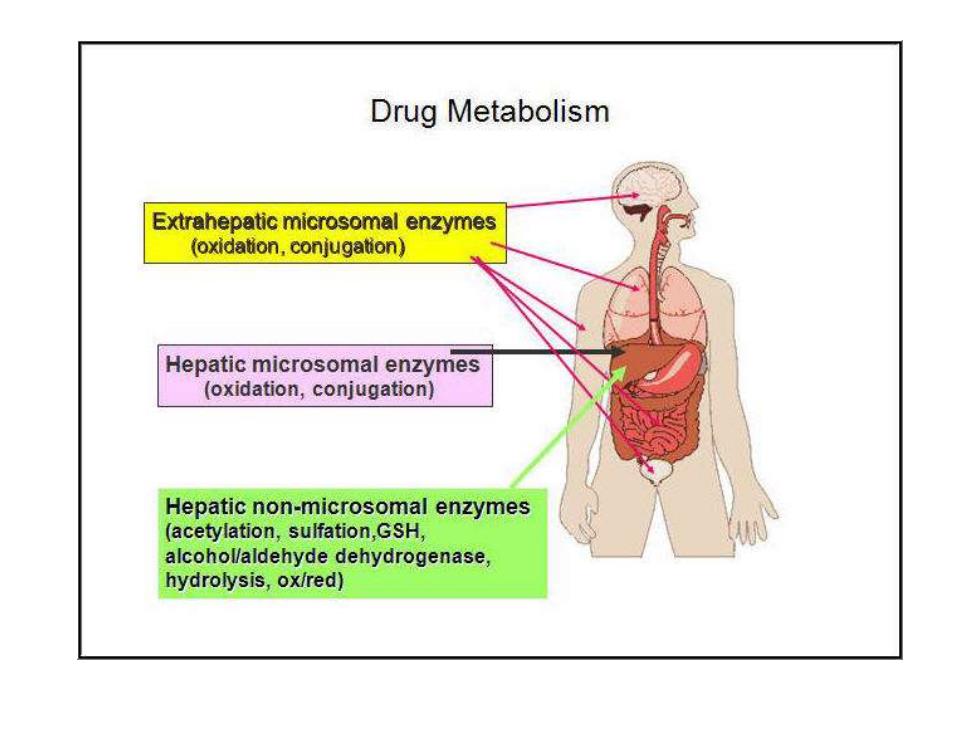
Drug Metabolism Extrahepatic microsomal enzymes (oxidation.conjugation) Hepatic microsomal enzymes (oxidation,conjugation) Hepatic non-microsomal enzymes (acetylation,sulfation,GSH, alcohollaldehyde dehydrogenase, hydrolysis,ox/red)】

药物作用的三个重要相 可供吸收药物 给药剂量 剂型崩解药物溶出 吸收 药剂相 生物利用度 药代动力相 失活产物 代谢失活 与蛋白 组织结合 结合药 游离药物 失活产物 代谢活化 排泄 转运形式 产生药理药性的 引起不良反应的 药效相 作用部位 作用部位 刺激 刺激 治疗效果 毒副反应
药物作用的三个重要相 给药剂量 剂型崩解药物溶出 产生药理药性的 作用部位 药剂相 药代动力相 药效相 吸收 可供吸收药物 生物利用度 与蛋白 结合药 物 游离药物 代谢失活 代谢活化 失活产物 失活产物 转运形式 组织结合 排泄 治疗效果 刺激 引起不良反应的 作用部位 毒副反应 刺激

药物的ADME 吸收:药物从给药部位进入循环系 统的过程 组织 f取决于药物理化性质 分在蛋白结合 肌肉或皮下注射 药 血被 静脉注射(f=100%) 作用部位 药物 (受体) 药理作 物 (游离型→ 结合型) 口服 消化道 肝 吸收 代谢 排泄 重吸收 肠胃道、皮下、 尿、胆汁 肾小管 肌肉等部位 肺等部位 肝肠循环 药剂相 药动相 药效相 Pharmaceutical Pharmacokinetic Pharmacodynemic phase phase phase
药物的ADME 吸收:药物从给药部位进入循环系 统的过程 (f=100%) f 取决于药物理化性质

常用参数 Characteristic Description Example value Abbreviation(s) Formula Dose Loading dose (LD),or steady state/maintenance dose(MD) 500mg D design parameter Dosing interval. 24h T design parameter The apparent volume in which a drug is distributed immediately after Volume of distribution it has been injected intravenously and equilibrated between plasma 6.0L =D/Co and the surrounding tissues. Concentration nitial or steady-state concentration of drug in plasma. 83.3μg/mL Co or Css =D/Va Biological halflife The time required for the concentration of the drug to reach half of its 12h original value. t12 In(2)/ke Elimination rate constant The rate at which drugs are removed from the body. 0.0578h1 In(2)/t2=CL/Va Elimination rate Rate of infusion required to balance elimination. 50 mg/h Kin =Css·CL The integral of the concentration-time curve (after a single dose or in AUCo-o Cdt Jo Area under the curve 1320μg/mLxh steady state). AUCTss rt+T Cdt Clearance The volume of plasma cleared of the drug per unit time. 0.38Lh CL = Va·ke=D/AUC AUCpo·D Bioavailability The fraction of drug that is absorbed. 0.8 f AUCn·Dpo Cmax The peak plasma concentration of a drug after oral administration 60.9μgmL Cmar direct measurement Time to reach Cmax. 3.9h direct measurement Cmin The lowest(trough)concentration that a drug reaches before the next 27.7μgfmL dose is administered. Cmin,ss direct measurement =100.Cmaz.ss--Cmm.s】 %PTF Cav,s Fluctuation Peak trough fluctuation within one dosing interval at steady state 41.8% where 生物利用度(bioavailability):制剂中药物被全身利用的程度,药物进入体内吸收后, 进入体循环的药量和给药量的比值,评价相同剂型吸收程度和疗效的重要指标。与脂溶 性和pKa相关
常用参数 生物利用度(bioavailability):制剂中药物被全身利用的程度,药物进入体内吸收后, 进入体循环的药量和给药量的比值,评价相同剂型吸收程度和疗效的重要指标。与脂溶 性和pKa相关