
二、新的测序策略-全基因组鸟枪法测序 对某基因组文库全部克隆片段进行末端序列 测定中未测到的碱基数,即缺口(gap),与已测定 的总碱基数相关。随着已测定碱基数的增加,缺 口的总碱基数目会按照泊松公式的一个推论 (P=e-m)迅速减小。其中P为基因组中某个碱基 未被测定的概率,m为所测定的碱基数与基因组 大小相比的倍数。m越大P值越小。当m值达到5 (即随机测定的碱基数达到基因组5倍时),基因 组中未测定的碱基数为基因组总碱基数的 0.67%(e-5=0.0067)。对流感嗜血杆菌这样大的基 因组(1.83Mb),可能留有128个平均长度为100bp 的缺口
二、新的测序策略-全基因组鸟枪法测序 对某基因组文库全部克隆片段进行末端序列 测定中未测到的碱基数,即缺口(gap),与已测定 的总碱基数相关。随着已测定碱基数的增加,缺 口的总碱基数目会按照泊松公式的一个推论 (P=e-m)迅速减小。其中P为基因组中某个碱基 未被测定的概率,m为所测定的碱基数与基因组 大小相比的倍数。m越大P值越小。当m值达到5 (即随机测定的碱基数达到基因组5倍时),基因 组中未测定的碱基数为基因组总碱基数的 0.67%(e-5=0.0067)。对流感嗜血杆菌这样大的基 因组(1.83Mb),可能留有128个平均长度为100bp 的缺口
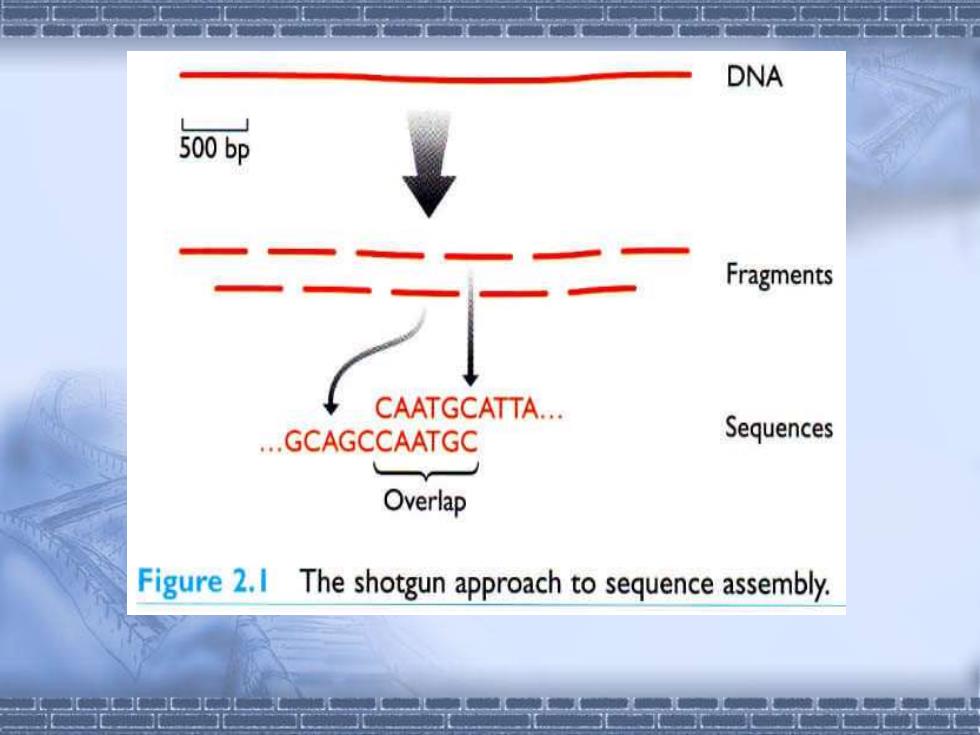
DNA 500bp Fragments CAATGCATTA. .GCAGCCAATGC Sequences Overlap Figure 2.1 The shotgun approach to sequence assembly

▪ 全基因组鸟枪法测序的主要步骤是: 第一,建立高度随机、插入片段大小为2kb左右的基因 组文库。克隆数要达到一定数量,即经末端测序的克隆片 段的碱基总数应达到基因组5倍以上。 第二,高效、大规模的末端测序。对文库中每一个克 隆,进行两端测序,TIGR在完成流感嗜血杆菌的基因组 时,使用了14台测序仪,用三个月时间完成了必需的 28,463个测序反应,测序总长度达6倍基因组。 第三,序列集合。TIGR发展了新的软件,修改了序列 集合规则以最大限度地排除错误的连锁匹配。 第四,填补缺口。有两种待填补的缺口,一是没有相 应模板DNA的物理缺口,二是有模板DNA但未测序的序列 缺口。他们建立了插入片段为15-20kb的λ文库以备缺口填 补
▪ 全基因组鸟枪法测序的主要步骤是: 第一,建立高度随机、插入片段大小为2kb左右的基因 组文库。克隆数要达到一定数量,即经末端测序的克隆片 段的碱基总数应达到基因组5倍以上。 第二,高效、大规模的末端测序。对文库中每一个克 隆,进行两端测序,TIGR在完成流感嗜血杆菌的基因组 时,使用了14台测序仪,用三个月时间完成了必需的 28,463个测序反应,测序总长度达6倍基因组。 第三,序列集合。TIGR发展了新的软件,修改了序列 集合规则以最大限度地排除错误的连锁匹配。 第四,填补缺口。有两种待填补的缺口,一是没有相 应模板DNA的物理缺口,二是有模板DNA但未测序的序列 缺口。他们建立了插入片段为15-20kb的λ文库以备缺口填 补
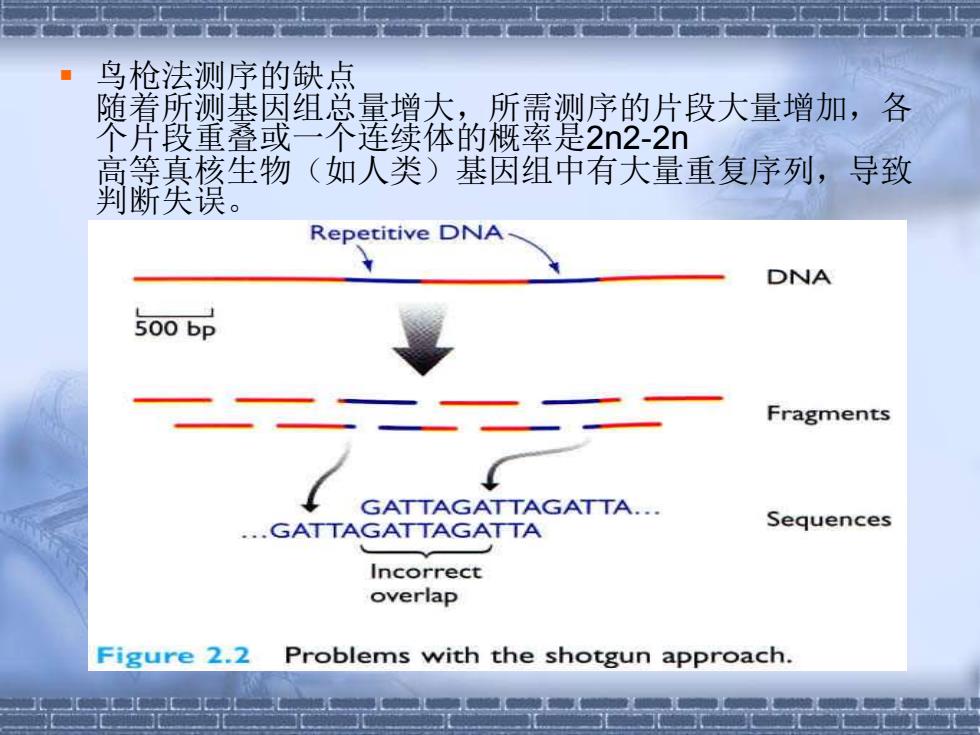
▪ 鸟枪法测序的缺点 随着所测基因组总量增大,所需测序的片段大量增加,各 个片段重叠或一个连续体的概率是2n2-2n 高等真核生物(如人类)基因组中有大量重复序列,导致 判断失误
▪ 鸟枪法测序的缺点 随着所测基因组总量增大,所需测序的片段大量增加,各 个片段重叠或一个连续体的概率是2n2-2n 高等真核生物(如人类)基因组中有大量重复序列,导致 判断失误

▪ 对鸟枪法的改进 (1) Clone contig法。 首先用稀有内切酶 把待测基因组降解 为数百kb以上的片 段,再分别测序。 (2) 靶标鸟枪法 (direted shotgun)。 首先根据染色体上 已知基因和标记的 位置来确定部分 DNA片段的相对位 置,再逐步缩小各 片段之间的缺口
▪ 对鸟枪法的改进 (1) Clone contig法。 首先用稀有内切酶 把待测基因组降解 为数百kb以上的片 段,再分别测序。 (2) 靶标鸟枪法 (direted shotgun)。 首先根据染色体上 已知基因和标记的 位置来确定部分 DNA片段的相对位 置,再逐步缩小各 片段之间的缺口