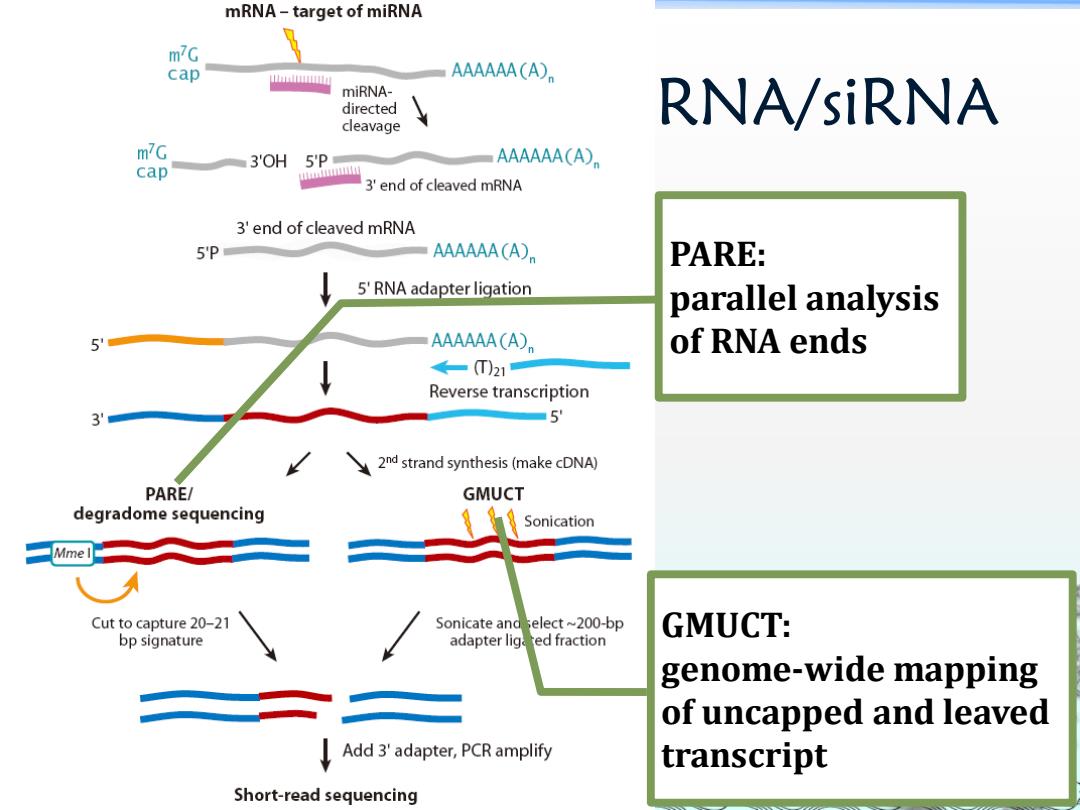
mRNA-target of miRNA m7G cap AAAAAACA) miRNA- directed RNA/siRNA cleavage m7G 3OH 5'p AAAAAA(A cap 3'end of cleaved mRNA 3'end of cleaved mRNA 5'P AAAAAACA) PARE: 5'RNA adapter ligation parallel analysis AAAAAA(A) of RNA ends 4一T)21 Reverse transcription 5 2nd strand synthesis(make cDNA) PARE/ GMUCT degradome sequencing Sonication Mme l Cut to capture 20-21 Sonicate and select~200-bp GMUCT: bp signature adapter lig ted fraction genome-wide mapping of uncapped and leaved Add 3'adapter,PCR amplify transcript Short-read sequencing
mRNA targets of miRNA/siRNA Analyses of mRNA targets of miRNA and mRNA degradation products by NGS methods. PARE: parallel analysis of RNA ends GMUCT: genome-wide mapping of uncapped and leaved transcript
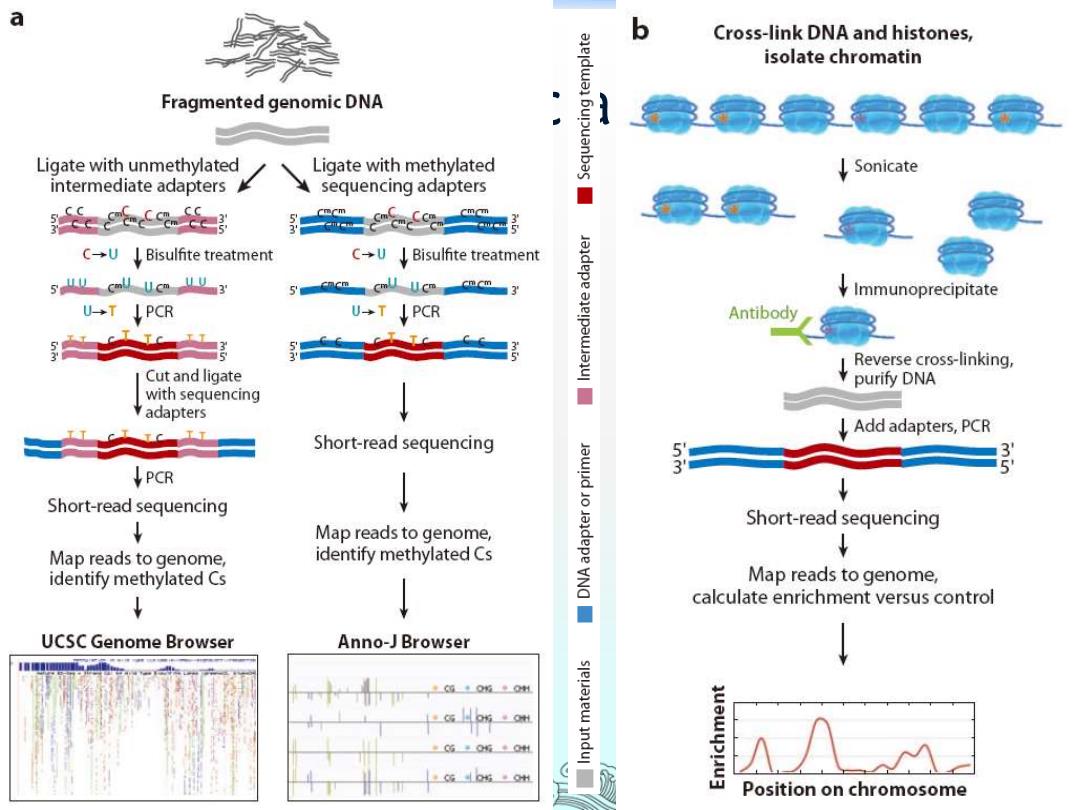
a b Cross-link DNA and histones, isolate chromatin Fragmented genomic DNA Ligate with unmethylated Ligate with methylated Sonicate intermediate adapters sequencing adapters ■ C→U↓Bisulfite treatment C→U↓Bisulfite treatment 5 uu Call Uca uU3 5cncm cmll u.cm cmcm 3 andepe ↓Immunoprecipitate U-T PCR U→T↓PCR Antibody 工1 Reverse cross-linking, Cut and ligate purify DNA with sequencing adapters ↓Add adapters,PcR Short-read sequencing 5 3 PCR Short-read sequencing ↓ Short-read sequencing Map reads to genome, Map reads to genome, identify methylated Cs identify methylated Cs Map reads to genome, ↓ ↓ calculate enrichment versus control UCSC Genome Browser Anno-J Browser 。G6年0+0 Position on chromosome
Epigenetic analyses

>based on genome sequence; >cross-hybridization (high background levels); >limited dynamic range of detection (<1000-fold); >normalization problems(across differentexperiments). ◇ Hybridization-based approache >based on expensive Sanger sequencing technology; >high throughput; >more precise; >a portion the short tags cannot be uniquely mapped. b) tag-based methods: CAGE:cap analysis of gene expression (~14-20 bp,5'ends) SAGE:serial analysis of gene expression (~14-20 bp,3'ends) MPSS:massively parallel signature sequencing (17-20 bp) Next-generation Sequencing-based method: RNA-Seq
RNA-Seq Technologies used in transcriptome studies: ✓ Hybridization-based approaches : a) Microarrays/chip; b) genomic tiling microarrays. ✓ Sequence-based approaches: a) EST: Expression Sequence Tag (~400 bp, 20-7000 bp) b) tag-based methods: ✓ CAGE: cap analysis of gene expression (~14-20 bp, 5′ ends) ✓ SAGE: serial analysis of gene expression (~14-20 bp, 3′ ends) ✓ MPSS: massively parallel signature sequencing (17-20 bp) ✓ Next-generation Sequencing-based method: RNA-Seq ➢based on genome sequence; ➢cross-hybridization (high background levels); ➢limited dynamic range of detection (<1000-fold); ➢normalization problems(across different experiments). ➢low throughput; ➢ expensive; ➢not quantitative. ➢based on expensive Sanger sequencing technology; ➢high throughput; ➢more precise; ➢a portion the short tags cannot be uniquely mapped

RNA-Seq Sequencing length:30-400bp. Advantages: > can be used to detect transcripts of any genome. low background,highly accurate large dynamic range of expression levels (~10000-fold) high levels of reproducibility (both for technical and biological replicates) requires less RNA sample (cloning steps) lower cost ◇
RNA-Seq Sequencing length: 30 - 400bp. Advantages: ➢ can be used to detect transcripts of any genome. ➢ low background, highly accurate ➢ large dynamic range of expression levels (~10000-fold) ➢ high levels of reproducibility (both for technical and biological replicates) ➢ requires less RNA sample (cloning steps) ➢ lower cost DNA sequencing technologies used for ranscriptome sequencing applications: ➢ Illumina IG, Applied Biosystems SOLiD and Roche 454 Life Science systems etc