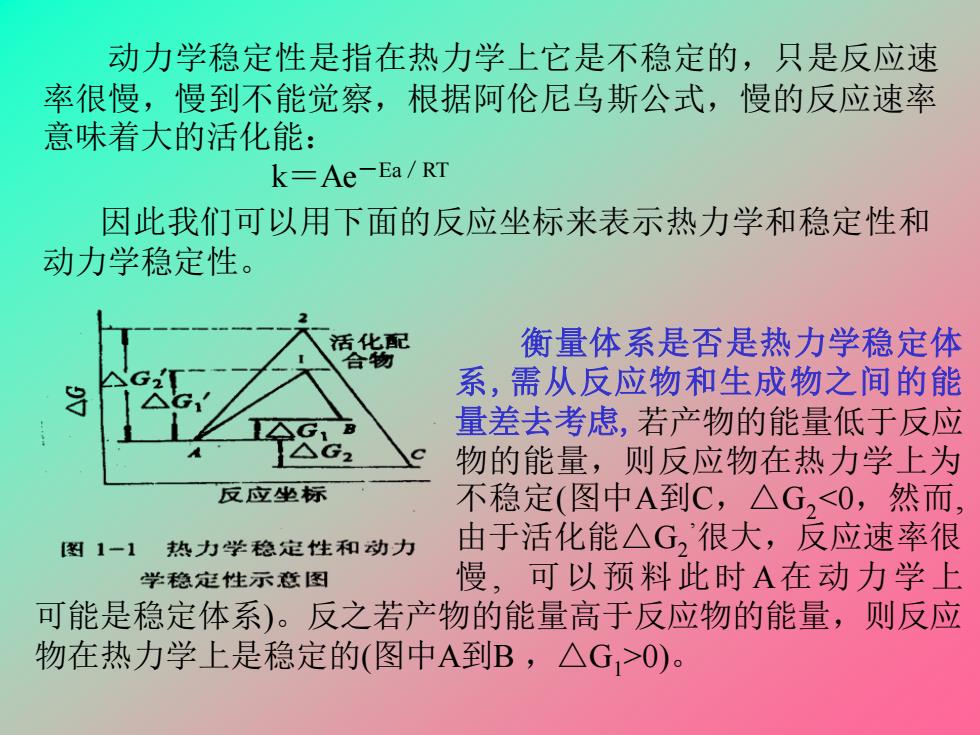
动力学稳定性是指在热力学上它是不稳定的,只是反应速 率很慢,慢到不能觉察,根据阿伦尼乌斯公式,慢的反应速率 意味着大的活化能: k=Ae-Ea/RT 因此我们可以用下面的反应坐标来表示热力学和稳定性和 动力学稳定性。 活化配 衡量体系是否是热力学稳定体 合物 系,需从反应物和生成物之间的能 量差去考虑,若产物的能量低于反应 物的能量,则反应物在热力学上为 反应坐标 不稳定(图中A到C,△G,<0,然而 图1一1热力学稳定性和动力 由于活化能△G,很大,反应速率很 学稳定性示意图 慢,可以预料此时A在动力学上 可能是稳定体系)。反之若产物的能量高于反应物的能量,则反应 物在热力学上是稳定的(图中A到B,△G,>0)
动力学稳定性是指在热力学上它是不稳定的,只是反应速 率很慢,慢到不能觉察,根据阿伦尼乌斯公式,慢的反应速率 意味着大的活化能: k=Ae-Ea/RT 衡量体系是否是热力学稳定体 系,需从反应物和生成物之间的能 量差去考虑,若产物的能量低于反应 物的能量,则反应物在热力学上为 不稳定(图中A到C,△G2<0,然而, 由于活化能△G2 ’很大,反应速率很 慢, 可以预料此时A在动力学上 可能是稳定体系)。反之若产物的能量高于反应物的能量,则反应 物在热力学上是稳定的(图中A到B ,△G1>0)。 因此我们可以用下面的反应坐标来表示热力学和稳定性和 动力学稳定性

动力学稳定性是从反应物与活化配合物之间的能量差,即反 应活化能来考虑的,若某一反应物转变为产物的趋势很大,反应 物在热力学上相当不稳定,但若实现这一反应所需的活化能也相 当大的话,则在动力学上反应物又是相当稳定的了。 在下面的图中,反应的自由焓变表明由A→B和由A→C都 是热力学不稳定体系,由于生成物C的能量更低,按理,从热力学 角度考虑将有利于反应A→C的进行,但是由于A→C的活化自由 能大于由A一B的活化自由能,所以从动力学角度考虑,将有利于 生成B。倘若△G,相当大,则完全有可能只生成B而不生成C。 热力学稳定性和动力学稳 活化配 合物 定性之间没有必然的联系。 催化剂可以改变反应的历程,亦即能改 变反应的活化能,因此,催化剂阿以增加或 降低动力学稳定性,但催化剂不能改变热 反应坐标 力学的稳定性或不稳定性。因为催化剂并 不改变反应的始态和终态,换言之,热力 图1-2 由动力学控制的热 学指出不能自发进行的反应是不能通过使 力学不稳定体系 用催化剂而使其发生的
动力学稳定性是从反应物与活化配合物之间的能量差,即反 应活化能来考虑的,若某一反应物转变为产物的趋势很大,反应 物在热力学上相当不稳定,但若实现这一反应所需的活化能也相 当大的话, 则在动力学上反应物又是相当稳定的了。 在下面的图中,反应的自由焓变表明由A B和由A C都 是热力学不稳定体系,由于生成物C的能量更低, 按理, 从热力学 角度考虑将有利于反应A C的进行, 但是由于A C的活化自由 能大于由A B的活化自由能, 所以从动力学角度考虑, 将有利于 生成B。倘若△G2 ’相当大,则完全有可能只生成B而不生成C。 热力学稳定性和动力学稳 定性之间没有必然的联系。 催化剂可以改变反应的历程,亦即能改 变反应的活化能,因此,催化剂可以增加或 降低动力学稳定性, 但催化剂不能改变热 力学的稳定性或不稳定性。因为催化剂并 不改变反应的始态和终态,换言之,热力 学指出不能自发进行的反应是不能通过使 用催化剂而使其发生的

般地,关于物质的稳定性,不外乎其本身是否容易分解, 该物质能否与环境中的某物种发生化学反应。所以在讨论物质的 稳定性时, 首先要列出该物质的所有可能的分解反应。如分解成单质、 分解成简单化合物、化合物的歧化: 其次还要考虑该物质是否能与大气中常见组分发生化学变化。 最后, 由各物质的标准生成自由焓算出一切可能的化学反应 的自由焓变。 与大气中组分的反应 物质 分解成单质 分解成简单化合物 △G=? 歧化 如果所有的可能反应的△G>O,就说该物质是稳定的;如果 有一个或多于1个反应的△G<0,即该物质对该反应是自发的,因 而该物质是不稳定的
一般地,关于物质的稳定性,不外乎其本身是否容易分解, 该物质能否与环境中的某物种发生化学反应。所以在讨论物质的 稳定性时, 首先要列出该物质的所有可能的分解反应。如分解成单质、 分解成简单化合物、化合物的歧化; 其次还要考虑该物质是否能与大气中常见组分发生化学变化。 最后,由各物质的标准生成自由焓算出一切可能的化学反应 的自由焓变。 如果所有的可能反应的△G>0,就说该物质是稳定的;如果 有一个或多于1个反应的△G< 0,即该物质对该反应是自发的,因 而该物质是不稳定的。 △G=? 与大气中组分的反应 分解成单质 分解成简单化合物 歧化 物质

如:讨论H,S的稳定性。 先查出它的△Gm=一34 kJmol-1 即H,S(g)=H,(g)+S(s)△Gn=34 kJ.mol-1 说明,S对于分解成单质来说是稳定的。 此处的标准生成自由焓,常常可以用来作为无机物相对于单 质的稳定性量度。如果△G<0,意味着由指定单质生成该物种 能量降低,生成反应是自发的,其逆反应的△G0,即分解为单 质是非自发的。这样,△G负值越大,化合物对于分解成单质稳 定性就越大。 不过在上述讨论中都是指的标准状态,然而 ①由于大气中的H,很少,其分压远小于1.01325×10Pa,且 △G的绝对值小于40,意指可以通过改变Q值而达到改变反应方 向。事实上,空气中H,的实际浓度为0.01%(V),计算得到的 △Gm≈21 kJmol-1,说明H,S在大气中对分解为单质是稳定的。 ②考虑H,S能与大气中的O,反应 HS+1/20,=H,O+S △G=-203.62<<-40kJmo- 所以对HS的稳定性的描述可以是这样: H,$在常温下大气中对于分解为单质是稳定的,但它能同氧发 生反应,所以H,S在大气中对氧化反应是不稳定的
如:讨论H2 S的稳定性。 先查出它的△fGm θ=-34 kJ·mol-1 即 H2 S (g)=H2 (g)+S(s) △rGm θ=34 kJ·mol-1 说明H2 S对于分解成单质来说是稳定的。 此处的标准生成自由焓,常常可以用来作为无机物相对于单 质的稳定性量度。如果△fGm θ<0,意味着由指定单质生成该物种 能量降低,生成反应是自发的,其逆反应的△Gθ>0,即分解为单 质是非自发的。这样, △fGm θ负值越大, 化合物对于分解成单质稳 定性就越大。 不过在上述讨论中都是指的标准状态,然而 ① 由于大气中的H2很少,其分压远小于1.01325×105Pa,且 △Gθ的绝对值小于40,意指可以通过改变Q值而达到改变反应方 向。事实上,空气中H2的实际浓度为0.01%(V),计算得到的 △rGm ≈ 21 kJ·mol-1 ,说明H2 S在大气中对分解为单质是稳定的。 ② 考虑H2 S能与大气中的O2反应 H2 S+1/2O2 =H2O+S △Gθ=-203.62 <<-40 kJ·mol-1 所以对H2 S的稳定性的描述可以是这样: H2 S在常温下大气中对于分解为单质是稳定的,但它能同氧发 生反应,所以H2 S在大气中对氧化反应是不稳定的

二吉布斯自由能变化与金属的还原 在无机化学中,认识金属氧化物、卤化物、硫化物的还原反应的规律十 分重要,而这种规律可以通过对还原反应进行热力学分析得到。 1氧化物的生成焓和还原反应的方向 不同的氧化物有不同的生成焓,为了能用热力学数据比较不 同氧化物的稳定性,首要的应消除不同价态造成的差别,习惯上 常按折合成消耗1molO,所形成的氧化物的生成焓进行比较,即: M+0,-3N0, △Hm(MO,) 如 3A1+02=3,0 △Hm(20,)=-1118 kJ-mol- 2Mh+02=2Mn,04 A H(MnO)=-694 kJ.mol-1
二 吉布斯自由能变化与金属的还原 在无机化学中,认识金属氧化物、卤化物、硫化物的还原反应的规律十 分重要,而这种规律可以通过对还原反应进行热力学分析得到。 1 氧化物的生成焓和还原反应的方向 不同的氧化物有不同的生成焓,为了能用热力学数据比较不 同氧化物的稳定性,首要的应消除不同价态造成的差别,习惯上 常按折合成消耗1mol O2所形成的氧化物的生成焓进行比较,即: M+O2 = MxOy △fHm θ ( MxOy ) 如 Al+O2 = Al2O3 △fHm θ ( Al2O3 )=-1118 kJ·mol-1 Mn+O2 = Mn3O4 △fHm θ ( Mn3O4 )=-694 kJ·mol-1 2x y 2 y 2 y 2 3 2 3 4 3 1 2 3 2 1 2