
心力小世】是指由于心功喷,以改心给出量减少,不足以活应全身组织代时需要的一种病理过程。心力有锡亦称东 可以包括两种情况:④心肌袁遇( 遇,此时系功能骑碍是原发的。如心肌炎时 心肌肌原纤维收缩功能得所数的 机的在 又如心肌梗死 杯死而致心肌收缩性减弱等等。②其他原 性相对不足而导致心力衰竭,此时泵功能得是继发的,在除去霾膜障碍时较易逆转。但心肌以外的原因3引起的心力衰,在晚期往往也伴 心肌损害,故在临床上有时两者不易区 ,有人把心脏受 包填塞)等原因引起的心脏泵血减少也包括在心力袁遇或泵衰遇的概念中。但是,由于引起心脏泵功 心力衰时,由于心输出量不能与辞回流相适应】 液可在系中。当心力贵是性经过时,往往有血容量和组织 ,并出现水 心力衰竭 (congestive heart failure) 急性心力衰竭多见于原发性心肌病变 侵性心力衰竭患老 也可发生急性心力 功能不全(c 质上是相同的,只是 程度上有所区 :心力袁 一般是指心功战 显的心力 两的 症材 ,在实际应用 中,这两个概念往往又是通用的, 第一节心力衰竭的原因和诱因 心力衰竭在临床上十分常见。它可既有心脏本身的疾病引起,也可继发于某些心外疾病如甲状腺功能亢进症,维生素B,缺乏等等。心力衰 调的病因可以摇括为下球三类: 一,心脏负荷加重 心脏的负荷可分为前负荷和后负荷两种,前负荷〔reload)或容量负荷是指心脏在收缩之前所承受的负荷,相当于心脏舒张末期的容量 前负荷的大小决定了心肌收缩的初长度,后负荷(coa)或压力负荷是指心整在收缩时所必须承受的负荷,相当于心腔壁在收缩时的张 力,但一般常以主动脉压作为左心室后负荷的指标。心脏负荷过重是心力责竭的常见原因。例如,在主动脉簿狡窄,高血压病或肺动脉高压 时,心脏后负荷过重,即压力负荷过重,即前负荷(容量负荷)过重.久之也能导致心力衰竭。在动静脉癀或严里苗血时,心输出量长期增 多,回心血量增多,故心脏所受的容量负荷也过重,因而他可引起心力贲蝎。在这种情况下,一股都先发生心肌吧大等代倦适应性变化,从而 使心功能可长期处于相对正常状态,最后则向代偿不全转化,而出现心力衰遇。 二、心肌代谢障碍 心肌对氧的需求量很大,必须有充分的血液和氧的供给才保精其正常功能.因此在严重或长朗的缺血缺氧时可发生心力意。心肌供 致的心肌供血相对不足可 他是引心力资提的因素之 此外严重的贫血和维生素B,缺乏,也可分别引起心肌供氧不足和生物氧化过程的隐碍,从而也可导致心力遇(详后文) 三、弥漫性心肌病 心肌炎,退行性心肌病等原发性心肌病变时,可因肌原纤维受到损去而使心肌收缩性减弱.如果损害严重或发展迅速,可导致急性心力盘 竭(如急性心肌炎时):若损害较轻,或病变呈慢性经过时,则对损害的反应是心肌,肥大等代偿话应性变化,因而在相当一段时间内心功能可 处于相对正常状态,但在一定条件下,如在某些诱因的作用下,代偿状态可转向代偿不全而发生心力衰竭。 心力袁蝎的诱因:促使心力衰姆发生的诱因很多。这些诱因基本上都是使心肌耗氧增加或供氧(供血)减少的因素,如感染(尤其是肺部 感染)、体力负荷过重、妊娠、分烧、情绪激动、心率过快或过漫、血压过高或一时性降低、输液过多等都可促使代偿失调而导致心力袁竭。 第二节心力衰竭的分类 心力衰竭的病因繁多,分类标准不一,常用的有以下几种分类法 一、根据心脏的受损部位分类 (一)左心衰竭主要是左心室搏出功能降碍,多见于冠状动脉粥样硬化性心脏病(尼心病)高血压病、主动脉雷狭窄或关闭不全、二尖酒 关闭不全等。机体的病理变化是由心输出量减少以及肺部淤血。水肿所引起。 (二)右心衰竭主要是右心室搏出功能障碍,见于肺心病、三尖鞭或肺动脉霾的疾病,并常继发于左心衰温。此时心输出量减少,体循环 淤血,静脉压增高,常伴有下肢水肿,严重时可发生全身性水钟。 (三)全心夜竭左、右心都发生衰竭称为全心衰竭,见于:@持久的左心衰渴可使右心负荷长期加重而导致右心衰竭:②心肌炎、心肌病 等病变如发生于全心、亦可引起全心衰竭。 二、根据发病的速度分类 (一)急性心力袁提发病急骤。心输出量急刷减少,机体来不及充分发挥代偿作用。常可伴有心源性休克。常见原因为急性心肌硬死,严 重的心肌炎等
心力衰竭(rhertfailure)是指由于心脏泵功能障碍,以致心输出量减少,不足以适应全身组织代谢需要的一种病理过程。心力衰竭亦称泵 衰竭(prmp failure)。在这一概念中可以包括两种情况:①心肌衰竭(myocardial failure):是指原发性心肌肌原纤维收缩功能障碍所致的心 力衰竭,此时泵功能障碍是原发的。如心肌炎时,心肌的变质、渗出或结缔组织增生可使心肌收缩性明显减弱。又如心肌梗死时可因部分心肌 坏死而致心肌收缩性减弱等等。②其他原因所致的心力衰竭:如心脏瓣膜病时,由于心肌负荷过重而发生心肌肥大和心脏扩大,继则心肌收缩 性相对不足而导致心力衰竭,此时泵功能障碍是继发的,在除去瓣膜障碍时较易逆转。但心肌以外的原因引起的心力衰竭,在晚期往往也伴有 心肌损害,故在临床上有时两者不易区分。 应当指出,有人把心脏受压(如心包填塞)等原因引起的心脏泵血减少也包括在心力衰竭或泵衰竭的概念中。但是,由于引起心脏泵功能 障碍最常见的原因是心肌收缩性减弱,故本章主要论述因原发性或继发性心肌收缩性减弱而发生的心力衰竭。 心力衰竭时,由于心输出量不能与静脉回流相适应,故血液可在静脉系统中淤积。当心力衰竭呈慢性经过时,往往伴有血容量和组织间液 的增多,并出现水肿,临床上称之为充血性心力衰竭(congestive heart failure)。急性心力衰竭多见于原发性心肌病变时,慢性心力衰竭患者 如心脏负荷突然加重,也可发生急性心力衰竭。 心功能不全(cardiacinsufficiency)与心力衰竭本质上是相同的,只是在程度上有所区别:心力衰竭一般是指心功能不全的晚期,患者有 明显的心力衰竭的临床症状,而心功能不全则指病情从轻到重的全过程,包括没有心力衰竭症状的心功能不全代偿阶段。但是,在实际应用 中,这两个概念往往又是通用的。 第一节 心力衰竭的原因和诱因 心力衰竭在临床上十分常见。它可既有心脏本身的疾病引起,也可继发于某些心外疾病如甲状腺功能亢进症,维生素B1缺乏等等。心力衰 竭的病因可以概括为下述三类: 一、心脏负荷加重 心脏的负荷可分为前负荷和后负荷两种,前负荷(preload)或容量负荷是指心脏在收缩之前所承受的负荷,相当于心脏舒张末期的容量, 前负荷的大小决定了心肌收缩的初长度,后负荷(afterload)或压力负荷是指心腔在收缩时所必须承受的负荷,相当于心腔壁在收缩时的张 力,但一般常以主动脉压作为左心室后负荷的指标。心脏负荷过重是心力衰竭的常见原因。例如,在主动脉瓣狭窄,高血压病或肺动脉高压 时,心脏后负荷过重,即压力负荷过重,即前负荷(容量负荷)过重、久之也能导致心力衰竭。在动静脉瘘或严重贫血时,心输出量长期增 多,回心血量增多,故心脏所受的容量负荷也过重,因而也可引起心力衰竭。在这种情况下,一般都先发生心肌肥大等代偿适应性变化,从而 使心功能可长期处于相对正常状态,最后则向代偿不全转化,而出现心力衰竭。 二、心肌代谢障碍 心肌对氧的需求量很大,必须有充分的血液和氧的供给才能保持其正常功能。因此在严重或长期的缺血、,缺氧时可发生心力衰竭。心肌供 血不足最常见的原因是冠状动脉粥样硬化。此时,由于冠脉血流量减少,病变部位心肌供血相对或绝对不足,故心肌收缩性可逐渐减弱而导致 心力衰竭。冠状动脉粥样硬化所引起的急性心肌梗死,也是心力衰竭的重要原因。在高血压病时,心肌代偿性肥大所致的心肌供血相对不足可 能也是引起心力衰竭的因素之一。 此外严重的贫血和维生素B1缺乏,也可分别引起心肌供氧不足和生物氧化过程的障碍,从而也可导致心力衰竭(详后文)。 三、弥漫性心肌病 心肌炎,退行性心肌病等原发性心肌病变时,可因肌原纤维受到损害而使心肌收缩性减弱。如果损害严重或发展迅速,可导致急性心力衰 竭(如急性心肌炎时);若损害较轻,或病变呈慢性经过时,则对损害的反应是心肌肥大等代偿适应性变化,因而在相当一段时间内心功能可 处于相对正常状态,但在一定条件下,如在某些诱因的作用下,代偿状态可转向代偿不全而发生心力衰竭。 心力衰竭的诱因:促使心力衰竭发生的诱因很多。这些诱因基本上都是使心肌耗氧增加或供氧(供血)减少的因素,如感染(尤其是肺部 感染)、体力负荷过重、妊娠、分娩、情绪激动、心率过快或过漫、血压过高或一时性降低、输液过多等都可促使代偿失调而导致心力衰竭。 第二节 心力衰竭的分类 心力衰竭的病因繁多,分类标准不一,常用的有以下几种分类法: 一、根据心脏的受损部位分类 (一)左心衰竭主要是左心室搏出功能障碍,多见于冠状动脉粥样硬化性心脏病(冠心病)高血压病、主动脉瓣狭窄或关闭不全、二尖瓣 关闭不全等。机体的病理变化是由心输出量减少以及肺部淤血、水肿所引起。 (二)右心衰竭主要是右心室搏出功能障碍,见于肺心病、三尖瓣或肺动脉瓣的疾病,并常继发于左心衰竭。此时心输出量减少,体循环 淤血,静脉压增高,常伴有下肢水肿,严重时可发生全身性水肿。 (三)全心衷竭左、右心都发生衰竭称为全心衰竭,见于:①持久的左心衰竭可使右心负荷长期加重而导致右心衰竭;②心肌炎、心肌病 等病变如发生于全心、亦可引起全心衰竭。 二、根据发病的速度分类 (一)急性心力衰竭发病急骤。心输出量急剧减少,机体来不及充分发挥代偿作用。常可伴有心源性休克。常见原因为急性心肌硬死,严 重的心肌炎等

(二)慢性心力袁竭较常见,病人长期处于一种持续的心力衰竭状态,并伴有静脉淤血和水肿。常见原因为心脏覆膜病、高血压病、肺动 脉高压等。 三、根据心力衰竭时心输出量的高低分类 (一)低心输出量性心力衰锡常见于冠心病、高血压病、心肌病、心脏舞要病等,此种病人在基础状态下心喻出量就低于正常。 (二)高输出量性心力衰竭继发于代谢增高或心脏后负荷降低的疾病如甲状腺机能亢进症、贫血、维生素B:缺乏病(脚气病)和动静脉 痿等。在此种情况下,由于循环血量增多或循环速度加快,心室前负荷增加,心输出量代偿性地增高,心脏必须作更多的功。但心肌的能量供 给却不足,故容易导致心力衰蝎。发生心力衰姆时心输出量比心力衰竭以前有所降低,但可稍高于正常水平,然而,由于组织需氧量增高、外 周血管扩张、动静脉短路等原因,这些病人的心输出量虽可比正常水平稍高。但组织的供氧量仍然不足。 第三节心功能不全发病过程中机体的代偿活动 心肌受损或心脏负荷加重时,体内出现一系列的代偿活动,通过这些代偿活动可使心血管系统的功能维持于相对正常状态。若病因维续作 用,则经过相当时间,在一定条件下代偿状态可以向失代偿状态转化而出现力心衰。 虽然机能代和形态的代是密切地相互联系的,但为了解的方使,可代分为以机能。代谢为主的代和以形态结构为主的代馈两 个方面 一、机能和代谢的代偿 正常人在运动或劳动时,才动员心血管系统的代偿活动。而心脏病患者则在基础情况下就需要动员这种代偿活动。能在短时间内被动员起 来的代偿活动 主要属于机能和代谢的代偿,有以下几种形式 (一)通过紧张源性扩张使心袖出量增加 正常心脏在回心血量增如,由于心室舒张末期容积及压力增加,心肌初长度增大,心室发生紧张源性扩张,按照Frank-Starling定律,此时 心肌收缩力加强,心输出量增加。心功能不全时,由于心泵功能减弱,心输出量减少,故心室舒张末期容积增加,心肌初长度增大:如肌节长 度不超过2.2μm,则这种紧张源性扩张也使心肌收缩力有所加强而起到代偿作用。 (仁)通过心交感神经和骨上腺蕾质释放儿茶酚胺增加使心肌收缩性加强。心率加快 心功能不全病人的交感神经系统活动加强。体力活动增加时血浆中去甲胃上腺素的含量比正常人有较明显的增多:24小时尿中去甲肾上腺 素的排出量也显著地高于正常人的排出量,说明心功能不全病人在休息时儿茶酚胺的分论也是增加的。交感肾上腺髓质系统的兴奋在维持功能 不全的心脏收缩性上起一定作用,另一方面也是心功能不全病人心率加快的基础, 一定程度的心率加快是最容易被迅速动员起来的一种代偿活动。正常人可通过心率加快使心输出量增加数倍.心功能不全时心率加快也县 一种重要的代偿形式,借此可使心输出量维持在一定的水平。但心率加快(如超过每分150一160次)时则由于心舒张期缩短,心肌耗氧量过 大,故每搏输出量明显减少,甚至因每分输出量减少而失去代偿意义, (但)心外的代偿 1,心输出量不足时交感肾上腺系统兴奋,外周小动脉的紧张性增加,有利于动脉血压维持在正常范国内。同时由于肾血流减少,肾素,血管 紧张素醛因酮系统被激活,从而导致体内钠、水藉留,使血容量增加,这对维持动款血正,也起一定作用。 2组织利用氧的能力增加心功能不全志者因血流变慢而发生箱环性缺氧。与此同时,组织。细胞中线粒体的呼吸酶活性增强,在慢性缺氧 时,细胞内线粒体的数量还可增多,因而组织利用氧的能力增强。 3.红细孢增多缺氧又可使血液细胞数和血红蛋白量增多。红细胞增多可提高血液携氧的能力,同时又有助于增加血量,故具有代偿意义。 二、形态结构的代偿·心肌肥大 在心脏负荷过重或心肌受损的初胡,首先出现的主要是机能和代谢的代偿,但与此同时也开给出现另一种代偿形式,即心肌形态结构的代 ,表现为心肌巴大 心肌大主要是心肌细胞体积增大的结果。心肌细胞一不发生增生,但有人报道,当心脏垂量招过500左右时,心肌纤维也可有数量的 增多.单位重量肥大心肌的收缩性是降低的,但由于整个心脏的重量增加,所以心脏总的收缩力加强,因此肥大心脏可以在相当长时间内处于 功能稳定状态,使每搏和每分输出量维持在话应机体需要的水平,使病人在相当长时间内不致发生心力竭。所以心肌大是心血管系统疾病 时起重要作用的二种 心肌肥大的发生机制至今尚未完全闸明。动物实 几秋内好可出现快速的话应姓反应表现为心肌收纯 加强。与此同时,核酸和蛋白质合成加强。主 窄后 天内心肌RN 含量可增加30 1g心肌中蛋白质含量每小时可增加1mg,所以 心肌总量很快增长 E ·关于 王机 如下的假设:当 时,心肌纤维的数量 P的 低,当心室受到过度的容量 负荷时,舒张 引起心肌纤维中 联性 心肌纤维 长度加大 心室腔因而扩大,即发 这两种配 代上君 里安TF用 过时,心肌肥大出现在心力袁竭之前。在相当长的时间内, 如数年甚至数十年内,心肌肥大可以代偿心脏的过度负 使心功能处于代偿粉段
(二)慢性心力衰竭较常见,病人长期处于一种持续的心力衰竭状态,并伴有静脉淤血和水肿。常见原因为心脏瓣膜病、高血压病、肺动 脉高压等。 三、根据心力衰竭时心输出量的高低分类 (一)低心输出量性心力衰竭 常见于冠心病、高血压病、心肌病、心脏瓣膜病等。此种病人在基础状态下心输出量就低于正常。 (二)高输出量性心力衰竭 继发于代谢增高或心脏后负荷降低的疾病如甲状腺机能亢进症、贫血、维生素B1缺乏病(脚气病)和动静脉 瘘等。在此种情况下,由于循环血量增多或循环速度加快,心室前负荷增加,心输出量代偿性地增高,心脏必须作更多的功。但心肌的能量供 给却不足,故容易导致心力衰竭。发生心力衰竭时心输出量比心力衰竭以前有所降低,但可稍高于正常水平。然而,由于组织需氧量增高、外 周血管扩张、动静脉短路等原因,这些病人的心输出量虽可比正常水平稍高。但组织的供氧量仍然不足。 第三节 心功能不全发病过程中机体的代偿活动 心肌受损或心脏负荷加重时,体内出现一系列的代偿活动,通过这些代偿活动可使心血管系统的功能维持于相对正常状态。若病因继续作 用,则经过相当时间,在一定条件下代偿状态可以向失代偿状态转化而出现力心衰竭。 虽然机能代谢和形态的代偿是密切地相互联系的,但为了理解的方便,可把代偿分为以机能、代谢为主的代偿和以形态结构为主的代偿两 个方面。 一、机能和代谢的代偿 正常人在运动或劳动时,才动员心血管系统的代偿活动。而心脏病患者则在基础情况下就需要动员这种代偿活动。能在短时间内被动员起 来的代偿活动,主要属于机能和代谢的代偿,有以下几种形式: (一)通过紧张源性扩张使心输出量增加 正常心脏在回心血量增加,由于心室舒张末期容积及压力增加,心肌初长度增大,心室发生紧张源性扩张,按照Frank-Starling定律,此时 心肌收缩力加强,心输出量增加。心功能不全时,由于心泵功能减弱,心输出量减少,故心室舒张末期容积增加,心肌初长度增大;如肌节长 度不超过2.2μm,则这种紧张源性扩张也使心肌收缩力有所加强而起到代偿作用。 (二)通过心交感神经和肾上腺髓质释放儿茶酚胺增加使心肌收缩性加强、心率加快 心功能不全病人的交感神经系统活动加强。体力活动增加时血浆中去甲肾上腺素的含量比正常人有较明显的增多;24小时尿中去甲肾上腺 素的排出量也显著地高于正常人的排出量,说明心功能不全病人在休息时儿茶酚胺的分泌也是增加的。交感-肾上腺髓质系统的兴奋在维持功能 不全的心脏收缩性上起一定作用,另一方面也是心功能不全病人心率加快的基础。 一定程度的心率加快是最容易被迅速动员起来的一种代偿活动。正常人可通过心率加快使心输出量增加数倍。心功能不全时心率加快也是 一种重要的代偿形式,借此可使心输出量维持在一定的水平。但心率加快(如超过每分150~160次)时则由于心舒张期缩短,心肌耗氧量过 大,故每搏输出量明显减少,甚至因每分输出量减少而失去代偿意义。 (三)心外的代偿 1.心输出量不足时交感-肾上腺系统兴奋,外周小动脉的紧张性增加,有利于动脉血压维持在正常范围内。同时由于肾血流减少,肾素-血管 紧张素-醛固酮系统被激活,从而导致体内钠、水潴留,使血容量增加,这对维持动脉血压,也起一定作用。 2.组织利用氧的能力增加心功能不全患者因血流变慢而发生循环性缺氧。与此同时,组织、细胞中线粒体的呼吸酶活性增强,在慢性缺氧 时,细胞内线粒体的数量还可增多,因而组织利用氧的能力增强。 3.红细胞增多缺氧又可使血液细胞数和血红蛋白量增多。红细胞增多可提高血液携氧的能力,同时又有助于增加血量,故具有代偿意义。 二、形态结构的代偿-心肌肥大 在心脏负荷过重或心肌受损的初期,首先出现的主要是机能和代谢的代偿,但与此同时也开始出现另一种代偿形式,即心肌形态结构的代 偿,表现为心肌肥大。 心肌肥大主要是心肌细胞体积增大的结果。心肌细胞一般不发生增生,但有人报道,当心脏重量超过500g左右时,心肌纤维也可有数量的 增多。单位重量肥大心肌的收缩性是降低的,但由于整个心脏的重量增加,所以心脏总的收缩力加强,因此肥大心脏可以在相当长时间内处于 功能稳定状态,使每搏和每分输出量维持在适应机体需要的水平,使病人在相当长时间内不致发生心力衰竭。所以心肌肥大是心血管系统疾病 时起重要作用的一种代偿形式。 心肌肥大的发生机制至今尚未完全阐明。动物实验表明,在实验性主动脉狭窄时,几秒钟内就可出现快速的适应性反应,表现为心肌收缩 加强。与此同时,核酸和蛋白质合成加强。主动脉狭窄后几天内心肌RNA含量可增加30~40%,1g心肌中蛋白质含量每小时可增加1mg,所以 心肌总量很快增长。 病理学家早就观察到,心肌肥大的两种形式:向心性肥大和远心性肥大(心脏扩大)。关于其发生机制,Grossman曾提出如下的假设:当 心室受到过度的压力负荷时,收缩期室壁压力的增高可引起心肌纤维中肌节的并联性增生,使心肌纤维变粗(在心脏总重量超过一定的监界值 时,心肌纤维的数量也可增多),室壁厚度增加,形成向心性肥大。这样,增厚了的心室壁可使收缩期室壁张力保持正常,使心输出量不致降 低。当心室受到过度的容量负荷时,舒张期室壁张力的增加可引起心肌纤维中肌节的串联性增生,心肌纤维长度加大,心室腔因而扩大,即发 生远心性肥大。这两种肥大在心脏功能的代偿上都起重要作用。 当心血管疾病呈慢性经过时,心肌肥大出现在心力衰竭之前。在相当长的时间内,如数年甚至数十年内,心肌肥大可以代偿心脏的过度负 荷或心肌损害,使心功能处于代偿阶段
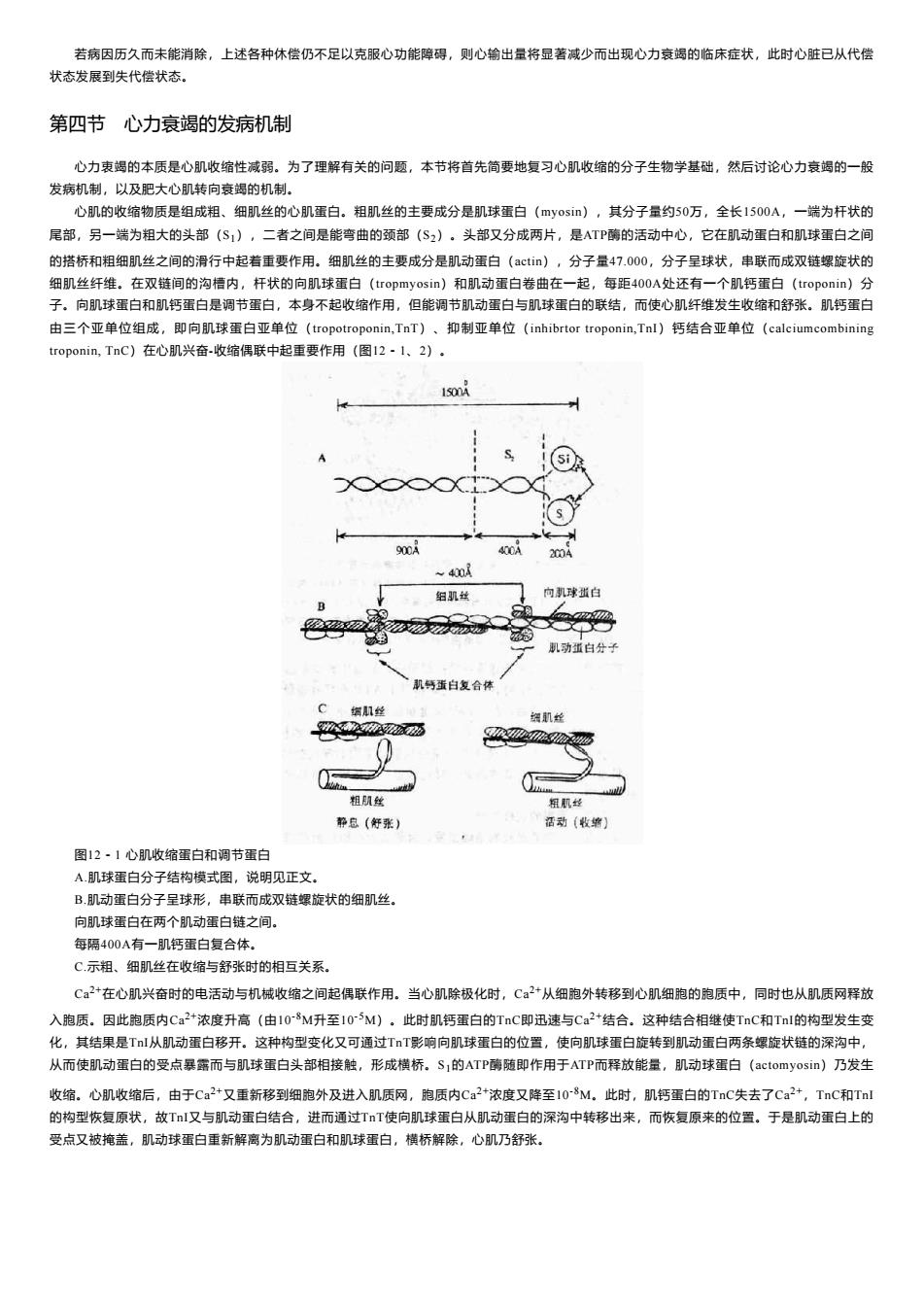
若病因历久而未能消除,上述各种休偿仍不足以克服心功能障碍,则心输出量将显著减少而出现心力衰竭的临床症状,此时心脏已从代圈 状态发展到失代偿状态。 第四节心力衰竭的发病机制 心力衷竭的本质是心肌收缩性减弱。为了理解有关的问题,本节将首先简要地复习心肌收缩的分子生物学基础,然后讨论心力衰竭的一般 发病机制,以及肥大心肌转向衰竭的机制, 心肌的收缩物质是组成粗、细肌丝的心肌蛋白,相肌丝的主要成分是肌球蛋白(my0s),其分子量约50万,全长1500A,一端为杆状的 尾部,另一端为相大的头部(S1),二者之间是能弯曲的颈部(S2)。头部又分成两片,是TP请的活动中心,它在肌动蛋白和肌球蛋白之间 的搭桥和闲细肌丝之司的行中起若重要作用.细肌丝的主要成分是肌动蛋白《c),分子量47000.分子里球状.率联而或双链螺旋状的 0A处 子向肌球蛋白和肌钙蛋白是调节蛋 本身不起 作用。但能调节肌动蛋白与肌球白的联结,而使肌纤维发生收和舒张肌号蛋 由三个亚单位组 即向肌 RI) 抑制亚单位 (inhibrtor tropo Tnl)钙结合亚单位(calcium troponin,TnC)在心肌兴奋-收缩偶联中起重要作用(图12 1.2) (si 组机 机号蛋白复合 粗机丝 形息(好张) 正动(最禁】 图12-1心肌收缩蛋白和调节蛋白 A肌球面白分子结构模式图,说明见正文。 B.肌动蛋白分子呈球形,串联而成双雠螺旋状的细肌丝。 向肌球蛋白在两个肌动蛋白链之间。 每原400A有一肌钙蛋白复合体, C示粗、细肌丝在数缩与舒张时的相互关系 C2在心肌兴奋时的电活动与机械收缩之间起偶联作用。当心肌除极化时,C2女从细胞外转移到心肌细胞的胞质中,同时也从肌质网释放 入胞质.因此胞质内Ca2浓度升高(由10-M升至10~5M),此时肌钙重白的TnC即迅速与Ca2*结合.这种结合相继使TnC和Tnl的构型发生变 化,其结果是T从肌动蛋白移开。这种构型变化又可通过TT影响向肌球蛋白的位置,使向肌球蛋白旋转到肌动蛋白两条螺旋状链的深沟中, 从而使肌动蛋白的受点暴露而与肌球蛋白头部相接触,形成祸桥。S,的ATP确随即作用于ATP而释放能量,肌动球蛋白(actomyosi1n)乃发生 由于C2“又重新移到细跑外及进入肌质网,胞质内C2浓度又降至10M此时,肌蛋白的TC失去了C,TC和 ,故Tn又与肌动蛋白 合,进而通过T 又拖”肌动白重离为动白和肌球白得伤院心机备 「使肌球蛋白从 1动蛋白的深沟中转移出来,而灰复原来的位置,于是肌动蛋白上的
若病因历久而未能消除,上述各种休偿仍不足以克服心功能障碍,则心输出量将显著减少而出现心力衰竭的临床症状,此时心脏已从代偿 状态发展到失代偿状态。 第四节 心力衰竭的发病机制 心力衷竭的本质是心肌收缩性减弱。为了理解有关的问题,本节将首先简要地复习心肌收缩的分子生物学基础,然后讨论心力衰竭的一般 发病机制,以及肥大心肌转向衰竭的机制。 心肌的收缩物质是组成粗、细肌丝的心肌蛋白。粗肌丝的主要成分是肌球蛋白(myosin),其分子量约50万,全长1500A,一端为杆状的 尾部,另一端为粗大的头部(S1),二者之间是能弯曲的颈部(S2)。头部又分成两片,是ATP酶的活动中心,它在肌动蛋白和肌球蛋白之间 的搭桥和粗细肌丝之间的滑行中起着重要作用。细肌丝的主要成分是肌动蛋白(actin),分子量47.000,分子呈球状,串联而成双链螺旋状的 细肌丝纤维。在双链间的沟槽内,杆状的向肌球蛋白(tropmyosin)和肌动蛋白卷曲在一起,每距400A处还有一个肌钙蛋白(troponin)分 子。向肌球蛋白和肌钙蛋白是调节蛋白,本身不起收缩作用,但能调节肌动蛋白与肌球蛋白的联结,而使心肌纤维发生收缩和舒张。肌钙蛋白 由三个亚单位组成,即向肌球蛋白亚单位(tropotroponin,TnT)、抑制亚单位(inhibrtor troponin,TnI)钙结合亚单位(calciumcombining troponin, TnC)在心肌兴奋-收缩偶联中起重要作用(图12-1、2)。 图12-1 心肌收缩蛋白和调节蛋白 A.肌球蛋白分子结构模式图,说明见正文。 B.肌动蛋白分子呈球形,串联而成双链螺旋状的细肌丝。 向肌球蛋白在两个肌动蛋白链之间。 每隔400A有一肌钙蛋白复合体。 C.示粗、细肌丝在收缩与舒张时的相互关系。 Ca 2+在心肌兴奋时的电活动与机械收缩之间起偶联作用。当心肌除极化时,Ca 2+从细胞外转移到心肌细胞的胞质中,同时也从肌质网释放 入胞质。因此胞质内Ca 2+浓度升高(由10 -8M升至10 -5M)。此时肌钙蛋白的TnC即迅速与Ca 2+结合。这种结合相继使TnC和TnI的构型发生变 化,其结果是TnI从肌动蛋白移开。这种构型变化又可通过TnT影响向肌球蛋白的位置,使向肌球蛋白旋转到肌动蛋白两条螺旋状链的深沟中, 从而使肌动蛋白的受点暴露而与肌球蛋白头部相接触,形成横桥。S1的ATP酶随即作用于ATP而释放能量,肌动球蛋白(actomyosin)乃发生 收缩。心肌收缩后,由于Ca 2+又重新移到细胞外及进入肌质网,胞质内Ca 2+浓度又降至10 -8M。此时,肌钙蛋白的TnC失去了Ca 2+,TnC和TnI 的构型恢复原状,故TnI又与肌动蛋白结合,进而通过TnT使向肌球蛋白从肌动蛋白的深沟中转移出来,而恢复原来的位置。于是肌动蛋白上的 受点又被掩盖,肌动球蛋白重新解离为肌动蛋白和肌球蛋白,横桥解除,心肌乃舒张

图12·2C2+与肌钙蛋白结合引起心肌收缩的示意图 在舒张期(左),肌钙重白复合体三成分(山,C、T)使向肌球蛋白 Tm】位于细肌丝(A》缓旋沟的外侧,从而阻止细肌丝与肌珠蛋白 楼桥(M)发生作用。当Ca2+与肌钙白C(右)结合引起一系列的 动使1与细肌丝分开,从而使Tm移进细肌丝 旋沟9 细肌丝乃得 以与肌球蛋白横桥相互作用而引起心肌收缩 皮目认为心A过重和心受装等行图3爬心农生 个方面 一、心肌能量代谢障碍 (一)能量生成(释放)障得 心肌主要借各种能源物质包括脂肪酸,葡葡糖等的有氧氧化而获得能量。心肌细跑对氧的需要量很大,摄取能力很强,在正常安静情况 下,冠状动静脉血氧含量差可高达14m%。可见,心肌氧供给不足或有氧氧化过程的障碍。均可使心肌细胞内能量生成不足而导致心肌收培性 减弱。 严重的贫血、冠状动脉硬化等所引起的心肌缺氧。是导致心肌细胞内能量生成不足的常见原因。维生素B,缺乏时,由于焦磷酸硫胺素(丙 酮酸脱羧璃的辅确)生成不足,丙酮酸的氧化发生避碍,故也可引起心肌能量生成不足。肥大的心肌也可因心肌缺氧而导致能量生成不足(详 后文) 一1结品田地周 心肌细胞内氧化磷酸化过程中所产生的TP,在心肌兴奋收缩偶联过程中受到肌球蛋白头部人TP裤的作用而水解,为心肌收缩提供能量。 实验表明,部分动物的心肌由肥大转向责蝎时,心肌托氧量和ATP含量并不减少而完成的机城功却显著减少,说明心肌利用ATP中的化学能作 机械功的过程有隐碍,即心肌的能量利用发生碍。有人发现,随着心肌负荷过重而发生心肌吧大时,心肌收缩蛋白的结构发生变化,肌球蛋 白头部ATP璃的活性降 ATP水铝发生 ,因此能量利用发生障碍,心肌收缩性乃因而减弱。这种现象也可见于老年人及甲状腺功能低下 的心。关于心肌收缩蛋白质结构发生变化的机制尚未闸明 二、兴奋-收缩偶联障碍-CaSB2+SB的运转失常 近年来。在心力衰提的发病机制中,因C+运转失常引起的心肌兴奋,收缩偶联隐碍,受到了很大重视,正常心肌在复极化时,心肌细胞内 肌质网的ATP南(钙泵)被激活,从而使胞质中的C✉2+逆着浓度差被摄取到肌质网中储存:同时,另一部分C2“则从胞质中被转运到细胞外。 于是心肌细胞胞质C✉2+浓度降低。心肌舒张。心肌除极化时,肌质网向胞质释放C✉+,同时又有C2从细胞外液进入胞质,因而胞质中C浓 度塔高,心肌收缩。心肌兴奋收缩偶联障碍的发生机制主要有:
图12-2 Ca 2+与肌钙蛋白结合引起心肌收缩的示意图 在舒张期(左),肌钙蛋白复合体三成分(I、C、T)使向肌球蛋白 (Tm)位于细肌丝(A)螺旋沟的外侧,从而阻止细肌丝与肌球蛋白 横桥(M)发生作用。当Ca 2+与肌钙蛋白C(右)结合引起一系列的 活动使I与细肌丝分开、从而使Tm移进细肌丝的螺旋沟中,细肌丝乃得 以与肌球蛋白横桥相互作用而引起心肌收缩。 下文将着重讨论慢性心力衰竭的发病机制。 从心肌分子结构及兴奋收缩偶联过程的基础出发,目前认为心肌负荷过重和心肌受损等病因引起心肌收缩性减弱的一般机制大致有下述几 个方面。 一、心肌能量代谢障碍 (一)能量生成(释放)障碍 心肌主要借各种能源物质包括脂肪酸,葡萄糖等的有氧氧化而获得能量。心肌细胞对氧的需要量很大,摄取能力很强,在正常安静情况 下,冠状动静脉血氧含量差可高达14ml%。可见,心肌氧供给不足或有氧氧化过程的障碍,均可使心肌细胞内能量生成不足而导致心肌收缩性 减弱。 严重的贫血、冠状动脉硬化等所引起的心肌缺氧,是导致心肌细胞内能量生成不足的常见原因。维生素B1缺乏时,由于焦磷酸硫胺素(丙 酮酸脱羧酶的辅酶)生成不足,丙酮酸的氧化发生障碍,故也可引起心肌能量生成不足。肥大的心肌也可因心肌缺氧而导致能量生成不足(详 后文)。 (二)能量利用障碍 心肌细胞内氧化磷酸化过程中所产生的ATP,在心肌兴奋-收缩偶联过程中受到肌球蛋白头部ATP酶的作用而水解,为心肌收缩提供能量。 实验表明,部分动物的心肌由肥大转向衰竭时,心肌耗氧量和ATP含量并不减少而完成的机械功却显著减少,说明心肌利用ATP中的化学能作 机械功的过程有障碍,即心肌的能量利用发生障碍。有人发现,随着心肌负荷过重而发生心肌肥大时,心肌收缩蛋白的结构发生变化,肌球蛋 白头部ATP酶的活性降低,ATP水解发生障碍,因此能量利用发生障碍,心肌收缩性乃因而减弱。这种现象也可见于老年人及甲状腺功能低下 的心脏。关于心肌收缩蛋白质结构发生变化的机制尚未阐明。 二、兴奋-收缩偶联障碍-Ca[SB]2+[/SB]的运转失常 近年来,在心力衰竭的发病机制中,因Ca 2+运转失常引起的心肌兴奋-收缩偶联障碍,受到了很大重视。正常心肌在复极化时,心肌细胞内 肌质网的ATP酶(钙泵)被激活,从而使胞质中的Ca 2+逆着浓度差被摄取到肌质网中储存;同时,另一部分Ca 2+则从胞质中被转运到细胞外。 于是心肌细胞胞质Ca 2+浓度降低。心肌舒张。心肌除极化时,肌质网向胞质释放Ca 2+,同时又有Ca 2+从细胞外液进入胞质,因而胞质中Ca 2+浓 度增高,心肌收缩。心肌兴奋-收缩偶联障碍的发生机制主要有:

,动球白 储能 结合 图12·3心肌能量代谢 (一)肌质网摄取Ca减少有人发现,在过度肥大的心肌中,肌质网ATP谤的活性降低,因而在心肌复极化时肌质网据取和储存Ca+的量 除极化时肌质网向胞质释放的C*也因之减少.由此所引起的心肌细胞除极化时胞质内C+浓度的低下可能是心肌收缩性减弱的重要原 另据报道,在肌质网摄取Ca+减少的同时,线粒体对Ca+摄取量增多,但线粒体在心肌除极化时向胞质程放Ca+的速度却非常缓慢。 C2*在心肌细胞中这种异常的分布也是胞质C2浓度降低的一个原因。此外,还有人认为线粒体内C2“的增多可引起氧化磷酸化脱偶联,从而 使能量生成不足 (二)酸中毒和高钾血症C的运转也受H和K的影响。在心力衰竭时有一定程度的缺氧,故可有细跑外液H和K浓度的蜡高。关于 H*如何影响运转的问题也未完全清楚。不同的作者损出过不同的假设:Kaz等曾认为H能在肌钙重白上与C2*竞争结合位置,因而在过多 时就能取代Ca2“的位置而使心肌的兴奋·收缩偶联发生障碍.也有人报道,在H浓度增高时,Ca2“与肌质网的结合比较牢固,除极化时肌质网 释放Ca2减少,故H增多时,心肌的兴奋收缩偶联发生障碍。前文已经提到,正常心肌细胞在除极化时有Ca2+从细胞外液进入胞质,而最近 有人报道,在细胞外液H浓度增高时,Ca的内流减慢,故心肌兴奋收缩偶联发生障得。 细胞外液中的K*和C2在心肌细胞上有互相竞争的作用。当外液中K+浓度升高时,动作电位中Ca2+内流就减少,因而心肌胞质中Ca2浓 应降低,这也是引起心肌兴奋收缩偶联障得的 个因素, (三)心肌内去甲肾上腺素含量减少有人报道,从心力衰漫患者取得的心房活组织中。去甲肾上腺素的含量很低:从有严重心力袁竭而行 二尖需置换术的志者取得的心室乳头肌活组织中,去甲肾上腺素的含量也很少,在有的病人,含量仅为正常的10%。心力袁竭时心肌去甲肾上 腺素含量减少,可能是由于肥大而意蝎的心肌中酪氨酸羟化酶活性降低,因而心肌交感神经纤维中儿茶酚胺的合成减少所致。在正常情况下, 去甲肾上镍素与心肌细跑表面的受体结合后,通过邀活豫苷酸环化酶,可使心肌细胞内的ATP转变为cAMPCCAMP一方面能促使Ca◆内流 另一方面又可通过蛋白激南的活化而使心肌细胞肌质网的一 种蛋白磷秀 化 摄取和释放Ca2+的速度增 可见去甲肾上豫素有州 的作 而心肌内去甲胃上腺素 含量 合成减少有关外,还可能是由于消耗过多。这是因为心输出量减少时 交感神经的活动加 ,故交神经 肌交 释放去甲胃上橡素增多 有些 时不仅有心肌U儿茶酚胺含量的减少 时.便丙基清上素作用时阳不产生正带量的cA心,损京可要E,病不足所所迷 而且心肌 能受体的功能也发生了改变 的不足就可使Ca+内流和肌质网摄取C2*不足,从而导致心肌兴奋-收缩偶联障得。 在这方面,也有一些不同的见解。有人认为,虽然心力衰竭时心肌儿茶酚胺含量减少,但因心力衰竭患者交感神经活动加强,故血液中儿 茶酚胺显著增多,而心肌对儿茶酚胺的敏感性也较高,故完全可能弥补心肌中儿茶酚胺的不足, 三、心肌的结构破坏 严重缺血时的心肌坏死,以及急性炎症时的心肌变性,坏死等可导致心肌收缩蛋白大量破坏,从而引起心肌收性显着减弱 关于肥大心肌转向 号的机制 前文已经有几外提到.为 的 心肌e大是 强有力 彩式然而它不是无限度的,如果病因历久 不能被消除, 大心肌的功能使不能长用维持正常而终转向 生大的基础上逐新发生发 把大的心肌为 期以来为人们进行探讨和研究的问 题。目前认为,代 大是一种不平衡的生长形式。这种在 器宜、组织细狗 、n (一)器 平 t 的增长超过 支配 脏的 神经分布的 ,肥大 儿茶酚胺合成 消耗增多 因而心内去甲上腺素含量显著减少, 速透色水平上的特征表场 会促使 联 目减少。对哺乳类动物心肌微猫环的活体组织研究表明,安静时,正常动物心肌每1mm内约有2,300条开放的毛细血管,毛细血管平均间距
图12-3 心肌能量代谢 (一)肌质网摄取Ca 2+减少有人发现,在过度肥大的心肌中,肌质网ATP酶的活性降低,因而在心肌复极化时肌质网摄取和储存Ca 2+的量 减少,除极化时肌质网向胞质释放的Ca 2+也因之减少。由此所引起的心肌细胞除极化时胞质内Ca 2+浓度的低下可能是心肌收缩性减弱的重要原 因。 另据报道,在肌质网摄取Ca 2+减少的同时,线粒体对Ca 2+摄取量增多,但线粒体在心肌除极化时向胞质释放Ca 2+的速度却非常缓慢。 Ca 2+在心肌细胞中这种异常的分布也是胞质Ca 2+浓度降低的一个原因。此外,还有人认为线粒体内Ca 2+的增多可引起氧化磷酸化脱偶联,从而 使能量生成不足。 (二)酸中毒和高钾血症 Ca 2+的运转也受H +和K +的影响。在心力衰竭时有一定程度的缺氧,故可有细胞外液H +和K +浓度的增高。关于 H +如何影响运转的问题也未完全清楚。不同的作者提出过不同的假设:Katz等曾认为H +能在肌钙蛋白上与Ca 2+竞争结合位置,因而在H +过多 时就能取代Ca 2+的位置而使心肌的兴奋-收缩偶联发生障碍。也有人报道,在H +浓度增高时,Ca 2+与肌质网的结合比较牢固,除极化时肌质网 释放Ca 2+减少,故H +增多时,心肌的兴奋-收缩偶联发生障碍。前文已经提到,正常心肌细胞在除极化时有Ca 2+从细胞外液进入胞质,而最近 有人报道,在细胞外液H +浓度增高时,Ca 2+的内流减慢,故心肌兴奋-收缩偶联发生障碍。 细胞外液中的K +和Ca 2+在心肌细胞上有互相竞争的作用。当外液中K +浓度升高时,动作电位中Ca 2+内流就减少,因而心肌胞质中Ca 2+浓 度降低,这也是引起心肌兴奋-收缩偶联障碍的一个因素。 (三)心肌内去甲肾上腺素含量减少有人报道,从心力衰竭患者取得的心房活组织中。去甲肾上腺素的含量很低:从有严重心力衰竭而行 二尖瓣置换术的患者取得的心室乳头肌活组织中,去甲肾上腺素的含量也很少,在有的病人,含量仅为正常的10%。心力衰竭时心肌去甲肾上 腺素含量减少,可能是由于肥大而衰竭的心肌中酪氨酸羟化酶活性降低,因而心肌交感神经纤维中儿茶酚胺的合成减少所致。在正常情况下, 去甲肾上腺素与心肌细胞表面的β受体结合后,通过激活腺苷酸环化酶,可使心肌细胞内的ATP转变为cAMP,cCAMP一方面能促使Ca 2+内流, 另一方面又可通过蛋白激酶的活化而使心肌细胞肌质网的一种蛋白磷酸化,从而使肌质网摄取和释放Ca 2+的速度增高。可见去甲肾上腺素有加 强心肌兴奋-收缩偶联的作用,而心肌内去甲肾上腺素含量减少时,心肌的兴奋-收缩偶联过程就可能发生障碍。肥大而衰竭的心肌内去甲肾上腺 素含量减少,除了可能与合成减少有关外,还可能是由于消耗过多。这是因为心输出量减少时,交感神经的活动加强,故交感神经末梢包括心 肌交感神经末梢释放去甲肾上腺素增多。 此外,有些事实还说明心力衰竭时不仅有心肌儿茶酚胺含量的减少,而且心肌细胞肾上腺能受体的功能也发生了改变,例如在严重心力衰 竭时,β受体受异丙基肾上腺素作用时细胞内不能产生正常量的cAM,提示可能此时β受体敏感性降低,因而cAMP产生不足。如前所述,cAMP 的不足就可使Ca 2+内流和肌质网摄取Ca 2+不足,从而导致心肌兴奋-收缩偶联障碍。 在这方面,也有一些不同的见解。有人认为,虽然心力衰竭时心肌儿茶酚胺含量减少,但因心力衰竭患者交感神经活动加强,故血液中儿 茶酚胺显著增多,而心肌对儿茶酚胺的敏感性也较高,故完全可能弥补心肌中儿茶酚胺的不足。 三、心肌的结构破坏 严重缺血时的心肌坏死,以及急性炎症时的心肌变性,坏死等可导致心肌收缩蛋白大量破坏,从而引起心肌收缩性显着减弱。 关于肥大心肌转向衰竭的机制,前文已经有几处提到。为了加深理解,下文将再就此问题作综合性的概述。 心肌肥大是一种强有力的代偿形式,然而它不是无限度的,如果病因历久而不能被消除,则肥大心肌的功能便不能长期维持正常而终转向 心力衰竭。慢性心力衰竭一般都是在心肌代偿性肥大的基础上逐渐发生发展的。 肥大的心肌为何会转向衰竭?这是长期以来为人们进行探讨和研究的问题。目前认为,代偿性心肌肥大是一种不平衡的生长形式。这种在 器官、组织、细胞、分子等不同的水平上都有其特征性表现的不平衡生长,是肥大心肌转向功能不全的基础。 (一)器官水平上的特征从整个心脏来看,不平衡生长表现为心脏重量的增长超过了支配心脏的交感神经元轴突的生长,因此心脏内交感 神经分布的密集程度显著地低于正常。而且,肥大心肌中儿茶酚胺合成减少而消耗增多,因而心内去甲肾上腺素含量显著减少,这种神经支配 和递质含量方面的变化,就会促使心肌兴奋-收缩偶联发生障碍,从而导致心肌收缩性减弱。 (二)组织水平上的特征表现在心肌内微动脉和毛细血管的生长明显地落后于心肌细胞体积的增长,所以单位重量的肥大心肌毛细血管数 目减少。对哺乳类动物心肌微循环的活体组织研究表明,安静时,正常动物心肌每1mm 3内约有2,300条开放的毛细血管,毛细血管平均间距