
benzene have been propo d CHAPTER 15 Benzene and Aromaticity Electrophilic Aromatic Substitution The ee ular structure of benzene is: 架 15-1 Naming the Benzenes 图 e is considered the "pa tic molecule. 上agmw 6-d ted benzenes are named using the prefixes en的ateeezenesaeohennamedbyaddngaprefix化 点 1
1 CHAPTER 15 Benzene and Aromaticity: Electrophilic Aromatic Substitution Benzene has the empirical formula CH. Its molecular formula has been shown to be C6H6, which implies four degrees of unsaturation in the molecule. Several incorrect structures of benzene have been proposed in the past: Of these, only Claus benzene has not been synthesized. All of the other structures represent unstable substances which isomerize to benzene in very exothermic reactions. The correct molecular structure of benzene is: Benzene is relatively inert. Among the reactions it does undergo is the reaction with bromine in the presence of catalytic amounts of FeBr3: The addition product is not formed, as might be expected. Further reaction with bromine leads to a mixture of isomeric di-substituted products: 15-1 Naming the Benzenes Benzene and its derivatives were originally called “aromatic compounds” because of their strong aromas. Benzene is considered the “parent” aromatic molecule. The structure of benzene is written as a pair of resonance structures or as a regular hexagon containing an inscribed circle: Monosubstituted benzenes are often named by adding a prefix to the word benzene: Disubstituted benzenes are named using the prefixes: •1,2- (ortho-, or o-) •1,3- (meta-, or m-) •1,4- (para-, or p-) The substituents are then listed in alphabetical order:

g benzene derivaivesllbeni o ames havebeen by UPA namv th um d its bas 空w之 ticity 8msoeSetmgna1n99ae,thean 2
2 For tri- and more highly substituted benzenes, the ring carbons are numbered to give the substituents the lowest set of numbers, as in cyclohexane nomenclature: The following benzene derivatives will be used often in this course: The naming of three substituted benzene systems do not follow IUPAC nomenclature in the Chemical Abstracts indexing preferences: phenol, benzaldehyde and benzoic acid. Ring substituted compounds of these substances are named by numbering the ring positions or using the prefixes o-, p- and m-. The carbon carrying the substituent giving the compound its base name is given the number 1. Several other common names have been accepted by IUPAC, including: A substituted benzene is called an arene. An arene, when used as a substituent, is called an aryl group (Ar). The parent aryl substituent is phenyl, C6H5-. The group C6H5CH2- is called phenylmethyl (benzyl). Structure and Resonance Energy of Benzene: a First Look at Aromaticity 15-2 At room temperature, benzene is inert to acids, H2, Br2 and KMnO4, reagents that usually add to conjugated alkenes. The cyclic 6-electron arrangement of double bonds imparts a special stability in the form of a large resonance energy

The benzene ring containssxequally overlapping p Byarogenssnecalwsable:heasod w ○ 6R。 《草心@ hstethamolecueont 15-3 Molecular Orbitals of Benzene 8ara8aastheenerovofbenaenea 1.3.5-hexatriene be compared to those 西 aromatic transitions 字望 eweg 3
3 The benzene ring contains six equally overlapping p orbitals. Experimentally, benzene can be shown to be a completely symmetrical hexagon; there are no alternating single (long) and double (short) bonds. The electronic structure of the benzene ring consists of 6 sp2 carbon atoms, with each p-orbital overlapping the p-orbitals of its nearest neighbors. Benzene is especially stable: heats of hydrogenation. It is useful to compare the heats of hydrogenation of benzene, 1,3-cyclohexadiene and cyclohexene. In each case, cyclohexane is the final product. The heat of hydrogenation of benzene can be experimentally measured using special catalysts and is measured to be 29.6 kcal mol-1 less than the calculated heat for the hypothetical molecule, 1,3,5-cyclohexatriene. Benzene is much more stable than a molecule containing alternating double and single bonds. The difference in stability is called the resonance energy of benzene, about 30 kcal mol-1. 15-3 π Molecular Orbitals of Benzene Cyclic overlap modifies the energy of benzene’s molecular orbitals. The molecular π orbitals of benzene can be compared to those of 1,3,5-hexatriene: The cyclic π system is stabilized relative to the acyclic one. Two of the orbitals have been lowered in energy in benzene (π1 and π3) while one has been raised (π2). The drop in energy of π1 and π3 more than offsets the rise in energy of π2. Some reactions have aromatic transitions states. The transition states in the Diels-Alder reaction, the addition of osmium tetroxide to alkenes and the first step in ozonolysis all exhibit a cyclic overlap of six electrons in π orbitals or orbitals having π character: These concerted aromatic transition states have lower energies than the alternative sequential bond-breaking and bond-making mechanism
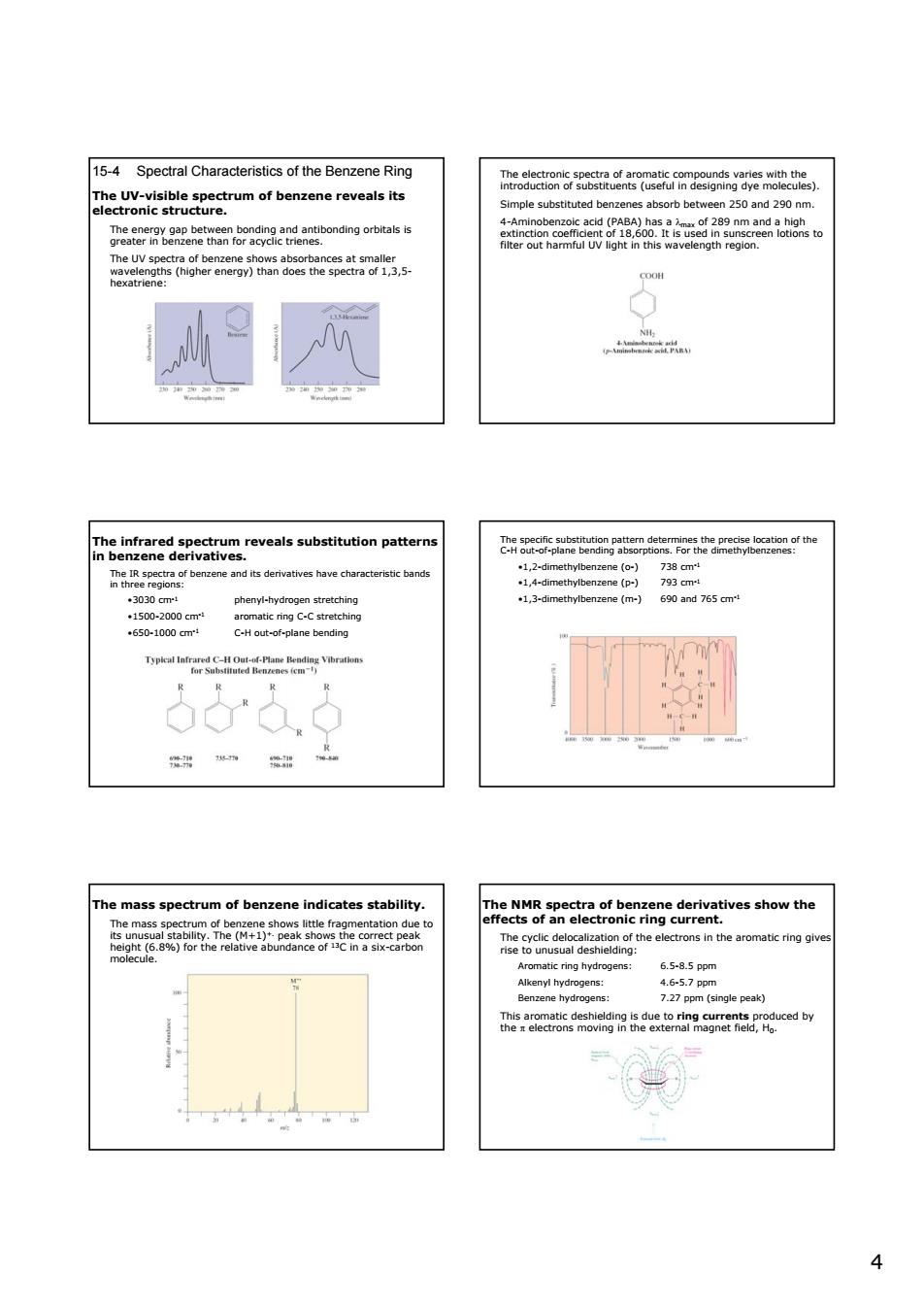
15-4 Spectral Characteristics of the Benzene Rine Teaeconkntasmecmegesgamethcte of benzene reveals t geaa82etwmn6a3e9dabcndngorbta teristic band phenyl-hydr 1,3-methybnene (m-) 690 and 765 om 650-1000cr: of ben ates tability he82Peac6heage.deravesshowhe ns in the aromatic ring ring h 6,5-85pp 4
4 15-4 Spectral Characteristics of the Benzene Ring The UV-visible spectrum of benzene reveals its electronic structure. The energy gap between bonding and antibonding orbitals is greater in benzene than for acyclic trienes. The UV spectra of benzene shows absorbances at smaller wavelengths (higher energy) than does the spectra of 1,3,5- hexatriene: The electronic spectra of aromatic compounds varies with the introduction of substituents (useful in designing dye molecules). Simple substituted benzenes absorb between 250 and 290 nm. 4-Aminobenzoic acid (PABA) has a λmax of 289 nm and a high extinction coefficient of 18,600. It is used in sunscreen lotions to filter out harmful UV light in this wavelength region. The infrared spectrum reveals substitution patterns in benzene derivatives. The IR spectra of benzene and its derivatives have characteristic bands in three regions: •3030 cm-1 phenyl-hydrogen stretching •1500-2000 cm-1 aromatic ring C-C stretching •650-1000 cm-1 C-H out-of-plane bending The specific substitution pattern determines the precise location of the C-H out-of-plane bending absorptions. For the dimethylbenzenes: •1,2-dimethylbenzene (o-) 738 cm-1 •1,4-dimethylbenzene (p-) 793 cm-1 •1,3-dimethylbenzene (m-) 690 and 765 cm-1 The mass spectrum of benzene indicates stability. The mass spectrum of benzene shows little fragmentation due to its unusual stability. The (M+1)+. peak shows the correct peak height (6.8%) for the relative abundance of 13C in a six-carbon molecule. The NMR spectra of benzene derivatives show the effects of an electronic ring current. The cyclic delocalization of the electrons in the aromatic ring gives rise to unusual deshielding: Aromatic ring hydrogens: 6.5-8.5 ppm Alkenyl hydrogens: 4.6-5.7 ppm Benzene hydrogens: 7.27 ppm (single peak) This aromatic deshielding is due to ring currents produced by the π electrons moving in the external magnet field, H0

bstituted ber theringand diminishes rapid (Benzene:7.27 ppm) hym have chemicash 1tmahPy2oaao2nhe2nhnteo6时rpe Ar Coepeg 32 15-5 Polycyclic Aromatic Hydrocarbons aled the ace Angular fusion (annulation")results in phenanthrene w8 5
5 The magnetic field from the ring current opposes H0 inside the loop but opposes it outside the loop where the hydrogens are located, resulting in deshielding. The effect is strongest closest to the ring and diminishes rapidly with distance. Benzylic nuclei are deshielded only about 0.4-0.8 ppm more than their allylic counterparts. Hydrogens farther away from π system have chemical shifts similar to those in the alkanes. Substituted benzenes may have more complicated NMR patterns. The presence of a substituent renders the ortho, meta and para hydrogens non-equivalent and subject to mutual coupling. (Benzene: 7.27 ppm) 4-(N,N-dimethylamino)benzaldehyde shows a large chemical shift difference between the two sets of ring hydrogens and a near first-order pattern of two doublets. The 9 Hz coupling constant is typical of splitting between ortho protons. All three types of coupling can be seen in the first order spectrum of 1-methoxy-2,4-dinitrobenzene (2,4-dinitroanisole). Ortho hydrogen (to methoxy) Doublet, δ=7.23 ppm, 9 Hz coupling Hydrogen flanked by nitro groups Doublet, δ=8.76 ppm, 3 Hz coupling Remaining ring hydrogen Doublet of doublets, δ=8.45 ppm, Para coupling between C3 and C6 is too small to be resolved. The 13C NMR spectra of benzene derivatives is not greatly affected by ring current shifts, since the induced ring current flows directly above and below the ring carbons. Benzene carbons exhibit chemical shifts similar to those in alkenes (120-135 ppm when unsubstituted). Benzene exhibits a single line at δ=128.7 ppm. 15-5 Polycyclic Aromatic Hydrocarbons Molecules containing several fused benzene rings are called polycyclic benzenoid or polycyclic aromatic hydrocarbons (PAHs). Common names are used for these systems, since there is no simple naming system for them. The series of linearly fused benzene rings is called the acenes. Angular fusion (“annulation”) results in phenanthrene. Quaternary carbons are numbered using the preceding carbon in the sequence followed by a letter indicating its distance to the preceding carbon