
第29卷第6期 催化学报 2008年6月 Vol.29 No.6 Chinese Journal of Catalysis June 2008 文章编号:0253-9837(2008)06-0524-07 研究论文:524~530 (Ag/Al2O3+Cu/Ce(x)/AI2O3)组合催化剂催化乙醇选择性 还原NOr及其副产物的消除 宋小萍,张长斌,贺泓 (中国科学院生态环境研究中心,北京100085) 摘要:制备了Cu/Ce(x)/Al2O3(x为Ce与Al的摩尔比)系列氧化催化剂,并考察了(Ag/Al2O3+Cu/Ce(x)/Al2O3)组合体系 催化乙醇还原NO以及氧化去除反应副产物(C0和未完全燃烧的碳氢化合物)的活性.在200一350℃温度区间,组合催化 剂具有与Ag/AI2O3相似的NO,去除效率.随着Ce/AI比增加,氧化催化剂去除CO的活性逐渐提高.Cu/CeO2催化剂具有最 好的氧化活性,但其对NO,的去除有较大影响.综合考虑NO,转化率以及CO和HC的去除效果,(Ag/Al2O3+Cu/Ce (0.15)/AI2O3)是最佳的催化剂组合体系.通过对此系列氧化催化剂的BET比表面积、XRD、H2-TPR以及XPS等表征结果的 分析,发现Cu和Ce之间的相互作用是催化剂氧化CO能力提高的主要原因. 关键词:银:氧化铝:氧化催化剂:氧化铜:氧化铺:氮氧化物:乙醇:选择性催化还原:一氧化碳:未燃碳氢化合物 中图分类号:0643 文献标识码:A Selective Catalytic Reduction of NO,by Ethanol and Removal of By- products over Combined Catalyst (Ag/Al2O3 Cu/Ce(x)/Al2O3) SONG Xiaoping,ZHANG Changbin,HE Hong" Research Center for Eco-Environmental Sciences,The Chinese Academy of Sciences,Beijing 100085,China) Abstract:The selective catalytic reduction of NO,by ethanol and the removal of by-products (CO and unburned hydrocarbons)were studied over (Ag/AlO3 Cu/Ce(x)/AlO3 )(x:molar ratio of Ce to Al)combined cata- lysts.The combined catalysts showed similar NO,reduction activity to Ag/Al2O3 in the temperature range of 200-350℃.CO oxidation capabilities of the oxidation catalysts were improved with increasing Ce/Al ratio. Cu/CeO,showed the best oxidation activity of CO but caused obvious decrease of NO,conversion when it was directly placed after Ag/Al2O3.(Ag/Al2O3 Cu/Ce(0.15)/Al2O3)was the best in the combined catalysts con- sidering the conversions of NO,,CO,and hydrocarbons.The Cu/Ce(x )/Al2O3 oxidation catalysts were charac- terized by BET surface area measurement,X-ray diffraction,H2 temperature-programmed reduction,and X-ray photoelectron spectroscopy.The results showed that the interaction between Cu and Ce is the main factor caus- ing the improvement of CO oxidation. Key words:silver:alumina:oxidation catalyst:copper oxide:cerium oxide:nitrogen oxide:ethanol:selective catalytic reduction:carbon monoxide:unburned hydrocarbons 催化碳氢选择性还原(HC-SCR)NO,技术是有原NO,的良好活性[1~31.其中Ag/AI2O3催化剂以 望应用于柴油发动机及稀燃发动机的尾气净化技术其良好的热稳定性、抗水性及抗硫性[45],成为最有 之一.许多催化剂表现出催化碳氢化合物(HC)还 望实际应用的HC-SCR催化剂体系之一.但研究表 收稿日期:2007-11-09、 联系人:贺泓,Tel:(010)62849123:Fax:(010)62923563:E-mail:honghe(@rcees.ac.cn. 基金来源:国家高技术研究发展计划(863计划,2006AA060304,2006AA06A304):国家重点基础研究发展计划(973计划, 2004CB719503)
文章编号:!"#$%&’$(("!!’)!)%!#"*%!( 研究论文:#"*!#$! 收稿日期:"!!(%++%!&, 联系人:贺 泓,-./:(!+!))"’*&+"$;012:(!+!))"&"$#)$;3%415/:67896.":;..<,1;,;8, 基金来源:国 家 高 技 术 研 究 发 展 计 划(’)$ 计 划,"!!)==!)!$!*,"!!)==!)=$!*);国 家 重 点 基 础 研 究 发 展 计 划(&($ 计 划, "!!*>?(+&#!$), (!"/!#$%&’()/(*(!)/!#$%&)组合催化剂催化乙醇选择性 还原 +%! 及其副产物的消除 宋小萍, 张长斌, 贺 泓 (中国科学院生态环境研究中心,北京 +!!!’#) 摘要:制备了 >@/>.(!)/=/"A$ (! 为 >.与 =/的摩尔比)系列氧化催化剂,并考察了(=9/=/"A$B>@/>.(!)/=/"A$)组合体系 催化乙醇还原 CA! 以及氧化去除反应副产物(>A和未完全燃烧的碳氢化合物)的活性,在"!!!$#! D温度区间,组合催化 剂具有与 =9/=/"A$ 相似的 CA! 去除效率,随着>./=/比增加,氧化催化剂去除>A的活性逐渐提高,>@/>.A" 催化剂具有最 好的氧化活性,但 其 对 CA! 的 去 除 有 较 大 影 响,综 合 考 虑 CA! 转 化 率 以 及 >A 和 E> 的 去 除 效 果,(=9/=/"A$B>@/>. (!F+#)/=/"A$)是最佳的催化剂组合体系,通过对此系列氧化催化剂的 ?3-比表面积、GHI、E"%-JH以及 GJK等表征结果的 分析,发现 >@和 >.之间的相互作用是催化剂氧化 >A能力提高的主要原因, 关键词:银;氧化铝;氧化催化剂;氧化铜;氧化铈;氮氧化物;乙醇;选择性催化还原;一氧化碳;未燃碳氢化合物 中图分类号:A)*$ 文献标识码:= ,*#*-./0*(1.1#2./-3*4)-./5657+%!829.:165#1643*;501#57<2= >?54)-.@50*?(5;8/6*4(1.1#2@.(!"/!#$%&’()/(*(!)/!#$%&) !"#$%&’()&*+,,-.#$/0’*+1&*,-2-(*+! ("#$#%&’()#*+#&,-&.’-/.*01&-*2#*+%34’1#*’#$,5(#)(1*#$#6’%7#28-,4’1#*’#$,9#1:1*;<===>?,)(1*%) !8@.?1-.:-6.<./.;L5M.;1L1/NL5;:.O@;L5787PCA!QN.L6187/18OL6.:.47M1/7PQN%R:7O@;L<(>A18O@8Q@:8.O 6NO:7;1:Q78<)S.:.<L@O5.O7M.:(=9/=/"A$B>@/>.(!)/=/"A$)(!:47/1::1L577P>.L7=/);74Q58.O;1L1% /N<L<,-6.;74Q58.O;1L1/N<L<<67S.O<545/1:CA!:.O@;L5781;L5M5LNL7=9/=/"A$58L6.L.4R.:1L@:.:189.7P "!!T$#! D,>A725O1L578;1R1Q5/5L5.<7PL6.725O1L578;1L1/N<L<S.:.54R:7M.OS5L658;:.1<589>./=/:1L57, >@/>.A"<67S.OL6.Q.<L725O1L5781;L5M5LN7P>AQ@L;1@<.O7QM57@<O.;:.1<.7PCA!;78M.:<578S6.85LS1< O5:.;L/NR/1;.O1PL.:=9/=/"A$,(=9/=/"A$B>@/>.(!F+#)/=/"A$)S1<L6.Q.<L58L6.;74Q58.O;1L1/N<L<;78% <5O.:589L6.;78M.:<578<7PCA!,>A,18O6NO:7;1:Q78<,-6.>@/>.(!)/=/"A$725O1L578;1L1/N<L<S.:.;61:1;% L.:5U.OQN?3-<@:P1;.1:.14.1<@:.4.8L,G%:1NO5PP:1;L578,E"L.4R.:1L@:.%R:79:144.O:.O@;L578,18OG%:1N R67L7./.;L:78<R.;L:7<;7RN,-6.:.<@/L<<67S.OL61LL6.58L.:1;L578Q.LS..8>@18O>.5<L6.4158P1;L7:;1@<% 589L6.54R:7M.4.8L7P>A725O1L578, A*2B5?4@:<5/M.:;1/@4581;725O1L578;1L1/N<L;;7RR.:725O.;;.:5@4725O.;85L:79.8725O.;.L6187/;<./.;L5M. ;1L1/NL5;:.O@;L578;;1:Q78478725O.;@8Q@:8.O6NO:7;1:Q78< 催化碳氢选择性还原(E>%K>H)CA! 技术是有 望应用于柴油发动机及稀燃发动机的尾气净化技术 之一,许多催化剂表现出催化碳氢化合物(E>)还 原 CA! 的良好活性[+!$] ,其中 =9/=/"A$ 催化剂以 其良好的热稳定性、抗水性及抗硫性[*,#],成为最有 望实际应用的 E>%K>H催化剂体系之一,但研究表 第"&卷 第)期 催 化 学 报 "!!’年)月 V7/,"&C7,) )(1*#$#@-A&*%3-,)%+%38$1$ W@8."!!’

第6期宋小萍等:(Ag/Al2O3+Cu/Ce(x)/Al2O3)组合催化剂催化乙醇选择性还原NOz及其副产物的消除 525 明,Ag/Al2O3催化剂在高效去除NO,的同时,不 将Ce作为添加成分引入到Al2O3载体中,研究了 可避免地会产生和N2生成量相当的CO6]和少量 (Ag/AlO3+Cu/Ce(x)/AlO3)组合催化剂体系去 未完全燃烧的乙醇和乙醛7:8]等反应副产物 除NO,以及CO的情况,并对优选出的最佳催化剂 在Ag/A山O3后放置氧化催化剂,将生成的CO 组合消除乙醇和乙醛的情况进行了考察.另外,采 以及未完全燃烧的HC氧化去除,是目前消除反应 用X射线衍射(XRD)、比表面积测定(BET)、氢气 过程中产生的副产物的普遍方法.Eranen等6]将商 程序升温还原(H2-TPR)以及X射线光电子能谱 用贵金属催化剂置于Ag/Al2O3之后,在150~600 (XPS)等表征手段对Cu/Ce(x)/Al,O3系列氧化催 ℃反应温度区间内,在尾气中未检测到CO生成,但 化剂进行了表征,探讨了Ce的量对催化剂氧化分解 催化体系去除NOz的活性大大降低.Miyadera试 CO活性的影响. 图以(Ag/Al,O3+CuSO,/TiO2+Pt/TiO2)组合催化 1实验部分 剂来达到在净化NO,的同时避免产生CO、乙醇、乙 醛、NH3及HCN等的目的.贵金属催化剂虽然在低 1.1催化剂的制备 温下即能将乙醇和乙醛完全氧化,但同时也容易将 Ag/Al2O3催化剂依文献[13]所述制备.贺泓 N2O,NH3,CHCN和HCN等氧化为NOz,从而 等13.141认为Ag/AlO3催化剂中Ag负载量为4% 影响体系去除NO,的效率.可见,后置氧化催化剂 时,NO,去除活性最佳.故文中Ag负载量定为 4%. 必须具有适中的氧化活性,在有效氧化CO及HC 的同时,不影响催化剂体系去除NO,的活性。 Cu/Ce(x)/Al2O3制备方法如下.按Ce/Al摩 尔比为0.01,0.06,0.15及0.30分别称取硝酸铈 张长斌等[8.]采用(Ag/Al2O3+Cu/Al2O3)组合 和Al2O3固体,溶于去离子水中,搅拌2h:将浆液 催化剂分别研究了乙醇和丙烯为还原剂的SCR反 旋转蒸发;将蒸干的固体置于烘箱中,于120℃烘 应中对NO,和产生的CO的消除情况,发现该组合 干过夜,再在500℃烧4h,得CeO2和Al2O3复合 催化剂体系具有与Ag/Al2O3相似的NO去除率, 载体,记为Ce(x)/A2O3(x=0.01,0.06,0.15, 但Cu/A12O3催化剂的低温氧化活性不强,直到350 0.30).称取Ce(x)/Al2O3复合载体溶于一定浓度 ℃(丙烯作还原剂)和400℃(乙醇作还原剂)才能分 的硝酸铜溶液中,其余步骤同前.所得催化剂记为 别达到对C0的100%去除,并且乙醇和乙醛也需要 Cu/Ce(x)/Al2O3(x=0.01,0.06,0.15,0.30). 在350℃才完全去除.另外,Cu/Al2O3氧化催化剂 Cu/CeO2催化剂按以下步骤制备:将硝酸铈于 与前置Ag/Al2O3采用1:1的堆体积比,大大增加了 450℃灼烧2h,制得CeO2固体:其余步骤同前. 组合催化剂体系的体积,限制了其实际应用.Koo- Cu/Al2O3的制备方法同文献[11,12].文中氧化催 va等[1o]以辛烷为还原剂,采用(Ag/Al,O3+Ag-H- 化剂均采用5%Cu负载量(以载体为基准). ZSM-5)组合催化剂(体积比1:1)去除NO,和CO, 1.2催化剂表征 发现组合催化剂去除NO,的低温活性有所提高,但 BET测试在美国Quantachrome公司的Quan- CO仍需在350℃才能完全去除.因此,如何在保持 tasorb-18型吸附分析仪上进行.于-196℃下,采 催化剂最紧凑组合的方式下,采用更少量的氧化催 用N2吸附容量法测定, 化剂,进一步降低CO和乙醇、乙醛的氧化温度,同 采用日本理学公司的D/MAX-RB型衍射仪鉴 时不影响催化剂体系对NO,的去除效率,成为后置 定催化剂的晶相结构,电压40kV,电流100mA, 氧化催化剂亟待解决的问题. CuK。射线(λ=0.154056nm),扫描20角的范围 CO2具有储氧和释放氧的功能,通常作为助剂 为10°-90°,扫描步长为4°/min. 组分存在于三效催化剂中,起到氧的缓冲作用. 催化剂的H2-TPR实验在内径为4mm的石英 CeO,作为载体能够与Cu或Ni等过渡金属活性组 管中进行,样品用量为200mg,筛分粒径为40~60 分相互作用,一方面提高活性组分在CO2载体表 目.脱附下来的气体用质谱(HPR20,Hiden Analyt- 面的分散度,另一方面活性组分也能促进CO2的 ic Ltd)检测质荷比(m/e)为2(H,)的信号,热电偶 还原112],从而提高催化剂的氧化活性 的温度信号和H2的质谱信号由电脑同步读取,得 基于前期的实验结果并考虑到Ce的特性,本文 到H2消耗峰随温度变化的TPR谱.具体步骤如
明,!"/!#$%& 催化剂在高效去除 ’%! 的同时,不 可避免地会产生和 ’$ 生成量相当的 (% [)]和少量 未完全燃烧的乙醇和乙醛[*,+]等反应副产物, 在 !"/!#$%& 后放置氧化催化剂,将生成的 (% 以及未完全燃烧的 -( 氧化去除,是目前消除反应 过程中产生的副产物的普遍方法,./0121等[)]将商 用贵金属催化剂置于 !"/!#$%& 之后,在345!)55 6反应温度区间内,在尾气中未检测到 (% 生成,但 催化体系去除 ’%! 的活性大大降低,789:;2/: [*]试 图以(!"/!#$%&<(=>%? /@8%$<AB/@8%$)组合催化 剂来达到在净化 ’%! 的同时避免产生(%、乙醇、乙 醛、’-& 及 -(’ 等的目的,贵金属催化剂虽然在低 温下即能将乙醇和乙醛完全氧化,但同时也容易将 ’$%,’-&,(-&(’ 和 -(’ 等氧化为 ’%!,从而 影响体系去除 ’%! 的效率,可见,后置氧化催化剂 必须具有适中的氧化活性,在有效氧化 (% 及 -( 的同时,不影响催化剂体系去除 ’%! 的活性, 张长斌等[+,C]采用(!"/!#$%&<(=/!#$%&)组合 催化剂分别研究了乙醇和丙烯为还原剂的 >(D 反 应中对 ’%! 和产生的 (%的消除情况,发现该组合 催化剂体系具有与 !"/!#$%& 相似的 ’%! 去除率, 但 (=/!#$%& 催化剂的低温氧化活性不强,直到&45 6(丙烯作还原剂)和?556(乙醇作还原剂)才能分 别达到对(%的355E去除,并且乙醇和乙醛也需要 在&45 6才完全去除,另外,(=/!#$%& 氧化催化剂 与前置!"/!#$%& 采用3F3的堆体积比,大大增加了 组合催化剂体系的体积,限制了其实际应用,GH1HI J:等[35]以辛烷为还原剂,采用(!"/!#$%&<!"I-I K>7I4)组合催化剂(体积比3F3)去除 ’%! 和 (%, 发现组合催化剂去除 ’%! 的低温活性有所提高,但 (%仍需在&45 6才能完全去除,因此,如何在保持 催化剂最紧凑组合的方式下,采用更少量的氧化催 化剂,进一步降低 (% 和乙醇、乙醛的氧化温度,同 时不影响催化剂体系对 ’%! 的去除效率,成为后置 氧化催化剂亟待解决的问题, (2%$ 具有储氧和释放氧的功能,通常作为助剂 组分 存 在 于 三 效 催 化 剂 中,起 到 氧 的 缓 冲 作 用, (2%$ 作为载体能够与 (=或 ’8等过渡金属活性组 分相互作用,一方面提高活性组分在 (2%$ 载体表 面的分散度,另一方面活性组分也能促进 (2%$ 的 还原[33,3$],从而提高催化剂的氧化活性, 基于前期的实验结果并考虑到(2的特性,本文 将(2作为添加成分引入到 !#$%& 载体中,研究了 (!"/!#$%&<(=/(2(!)/!#$%&)组合催化剂体系去 除 ’%! 以及 (%的情况,并对优选出的最佳催化剂 组合消除乙醇和乙醛的情况进行了考察,另外,采 用 L射线衍射(LDM)、比表面积测定(N.@)、氢气 程序升 温 还 原(-$I@AD)以 及 L 射 线 光 电 子 能 谱 (LA>)等表征手段对 (=/(2(!)/!#$%& 系列氧化催 化剂进行了表征,探讨了(2的量对催化剂氧化分解 (%活性的影响, ! 实验部分 !"! 催化剂的制备 !"/!#$%& 催化剂依文献[3&]所述制备,贺泓 等[3&,3?]认为 !"/!#$%& 催化剂中 !"负载量为?E 时,’%! 去 除 活 性 最 佳,故 文 中 !" 负 载 量 定 为 ?E, (=/(2(!)/!#$%& 制备方法如下,按 (2/!#摩 尔比为5O53,5O5),5O34及5O&5分别称取硝酸铈 和 !#$%& 固体,溶于去离子水中,搅拌$P;将浆液 旋转蒸发;将蒸干的固体置于烘箱中,于3$5 6烘 干过夜,再在455 6烧 ?P,得 (2%$ 和 !#$%& 复合 载体,记 为 (2(!)/!#$%& (!Q5O53,5O5),5O34, 5O&5),称取 (2(!)/!#$%& 复合载体溶于一定浓度 的硝酸铜溶液中,其余步骤同前,所得催化剂记为 (=/(2(!)/!#$%& (!Q5O53,5O5),5O34,5O&5), (=/(2%$ 催化剂按以下步骤制备:将硝酸铈于 ?456灼烧 $P,制得 (2%$ 固体;其余步骤同前, (=/!#$%& 的制备方法同文献[33,3$],文中氧化催 化剂均采用4E(=负载量(以载体为基准), !"# 催化剂表征 N.@ 测试在美国 R=:1B:SP/HT2公司的 R=:1I B:UH/VI3+型吸附分析仪上进行,于W3C) 6 下,采 用 ’$ 吸附容量法测定, 采用日本理学公司的 M/7!LIDN型衍射仪鉴 定催化剂的晶相结构,电压?5XY,电流355T!, (="! 射线("Q5O34?54)1T),扫描$#角的范围 为35Z!C5Z,扫描步长为?Z/T81, 催化剂的 -$I@AD实验在内径为 ?TT 的石英 管中进行,样品用量为$55T",筛分粒径为?5!)5 目,脱附下来的气体用质谱(-AD$5,-8;21!1:#9BI 8S[B;)检测质荷比(#/$)为$(-$)的信号,热电偶 的温度信号和 -$ 的质谱信号由电脑同步读取,得 到 -$ 消耗峰随温度变化的 @AD 谱,具体步骤如 第)期 宋小萍 等:(!"/!#$%&<(=/(2(!)/!#$%&)组合催化剂催化乙醇选择性还原 ’%! 及其副产物的消除 4$4

526 催化学报 第29卷 下:首先将200mg样品在500℃,30ml/min的 20%O2/He气氛中预处理1h,然后冷至室温:室 。CeO2 温下通He吹扫1h,切换为30ml/min的5%H,/ y-Al.O3 Ar,并以l0℃/min的升温速率进行TPR实验. XPS实验采用ESCALAB MarkⅡ能谱仪(英国 (6) VG公司)测定.A1K。=1486.6eV为激发光源,采 用C1s=285.0eV校正荷电位移. (5) 1.3催化剂活性评价 (4) 活性评价在由计算机控制的六气路固定床反应 (3) 装置上进行.取平均粒径20~40目的催化剂进行 (2) 稳态实验,反应温度为150~600℃.反应样气模拟 (1) 柴油机尾气组成:p(NO)=0.08%,9(O2)= 10 20 30 40 5060 7080 90 10%,(C0)=0.06%,p(C2HOH)=0.1565%, 28/(°) (H,O)=10%,N2作为平衡气:反应气流速为 图1Cu/Ce(x)/Al,O3氧化催化剂的XRD谱 2L/min.所有原料气均为钢瓶气,经过配气系统均 Fig 1 XRD patterns of the Cu/Ce(x)/AlO;oxidation catalysts (x=Ce/Al molar ratio) 匀混合后进入反应管.乙醇及水的添加利用注射器 (1)Cu/ALO3,(2)Cu/Ce(0.01)/AlO3 泵和汽化炉精确控制液体的蒸发量并随载气带入反 (3)Cu/Ce(0.06)/AlO3,(4)Cu/Ce(0.15)/Al2O, 应器.只考虑氧化催化剂与前置Ag/A2O3催化剂 (5)Cu/Ce(0.30)/Al2O3·(6)Cu/CeO2 紧密放置的情况,组合催化剂记为(Ag/Al2O3+Cu/ 大.Cu/CeO2具有最小的比表面积.Ce添加对Cu/ Ce(x)/A2O3),Ag/Al2O3堆体积为Cu/Ce(x)/ A2O3比表面积的影响机理和变化规律还有待深入 Al2O3的3倍.NO,的测定采用美国Monitor Labs 研究.结合此后的活性数据发现,催化剂活性与比 公司9841型NO,化学发光仪,CO的测定采用美 表面积之间并没有线性关系,因此我们认为比表面 国Agilent6890型气相色谱仪(TCD检测器,5A分 积并不是影响催化剂活性的主要因素. 子筛填充柱),乙醇和乙醛采用GCMS联用技术 表1Cu/Ce(x)/A山,O3系列氧化催化剂的比表面积 (美国Agilent6890N-5973NGC-MS)分析. Table 1 Surface area of the oxidation catalysts of Cu/Ce()/AlOs 2结果与讨论 Catalyst BET surface area (m2/g) Cu/Al2O; 2.1催化剂的表征 229.2 Cu/Ce(0.01)/Al2O 178.9 2.1.1XRD和BET表征结果 Cu/Ce(0.06)/Al2O3 237.6 对Cu/Ce(x)/Al2O3系列氧化催化剂进行了 Cu/Ce(0.15)/AlO 328.4 XRD和BET表征,结果分别示于图1和表1.由图 Cu/Ce(0.30)/Al2O; 160.6 Cu/CeO2 85.3 1可见,Cu/Al2O3上出现了Y-Al2O3衍射峰.随Ce 的添加,载体y-Al2O3的衍射峰消失,Ce的添加破坏 为了研究Ce的添加使Cu/Ce(x)/Al2O3系列 了部分Y-Al2O3的晶形结构,使其以无定形态存在. 氧化催化剂活性提高的原因,下面进一步对Cu/Ce Cu/Ce(x)/Al2O3以及Cu/CeO2氧化催化剂上存在 (x)/Al2O3系列催化剂进行H2-TPR及XPS表征. 的主要结晶相为CeO2萤石相,其衍射峰强度随Ce/ 2.1.2H2-TPR表征结果 A1比的增加逐渐增强.氧化催化剂上未出现CuO 图2是Cu/Ce(x)/Al2O3催化剂的H2-TPR 的衍射峰,这说明活性组分Cu可能主要以高度分 谱,可见随着Ce/Al比变化,催化剂的H2-TPR谱发 散的状态存在15] 生了显著的变化.Cu/Al2O3在271℃和302℃出现 由表1中的结果可见,少量Ce(Ce/Al=0.01) 了两个还原峰,分别记为a和B.随着Ce的添加,a 的添加显著降低了Cu/Al2O3催化剂的比表面积. 峰消失,B峰逐渐增大并向低温移动,当Ce/Al比 而当Ce添加量进一步升高,催化剂比表面积先增大 大于0.06时,y峰开始出现.Ce/A1比为0.30时, 后减小.当Ce/A1比为0.15时,比表面积达到最 出现了新峰6
下:首先将 !"" #$样品在 %"" &,’" #(/#)*的 !"+ ,! /-.气氛中预处理 /0,然后冷至室温;室 温下通 -.吹扫 /0,切换为’"#(/#)*的%+-! / 12,并以/" &/#)*的升温速率进行 345实验6 748实验采用98:1;1<=>2?!能谱仪(英国 @A 公司)测定61(!!B/CDEFE.@ 为激发光源,采 用 :/"B!D%F".@ 校正荷电位移6 !"# 催化剂活性评价 活性评价在由计算机控制的六气路固定床反应 装置上进行6取平均粒径!""C"目的催化剂进行 稳态实验,反应温度为/%""E"" &6反应样气模拟 柴 油 机 尾 气 组 成:"(G,)B"F"D+,"(,!)B /"+,"(:,)B"F"E+,"(:!-%,-)B"F/%E%+, "(-!,)B/"+,G! 作 为 平 衡 气;反 应 气 流 速 为 !;/#)*6所有原料气均为钢瓶气,经过配气系统均 匀混合后进入反应管6乙醇及水的添加利用注射器 泵和汽化炉精确控制液体的蒸发量并随载气带入反 应器6只考虑氧化催化剂与前置 1$/1(!,’ 催化剂 紧密放置的情况,组合催化剂记为(1$/1(!,’H:I/ :.(#)/1(!,’),1$/1(!,’ 堆 体 积 为 :I/:.(#)/ 1(!,’ 的’倍6G,# 的测定采用美国 =J*)KJ2;>LM 公司NDC/型 G,# 化学发光仪,:, 的测定采用美 国 1$)(.*KEDN"型气相色谱仪(3:O 检测器,%1 分 子筛填充柱),乙醇和乙醛采用 A:P=8 联用技术 (美国 1$)(.*KEDN"GP%NQ’GA:P=8)分析6 图 ! $%/$&(!)/’()*# 氧化催化剂的 +,-谱 R)$/ 75OS>KK.2*MJTK0.:I/:.(#)/1(!,’JU)V>K)J* W>K>(XMKM(#B:./1(#J(>22>K)J) (/):I/1(!,’,(!):I/:.("F"/)/1(!,’, (’):I/:.("F"E)/1(!,’,(C):I/:.("F/%)/1(!,’, (%):I/:.("F’")/1(!,’,(E):I/:.,! ) 结果与讨论 )"! 催化剂的表征 )"!"! +,-和 ./0表征结果 对 :I/:.(#)/1(!,’ 系列氧化催化剂进 行 了 75O和<93表征,结果分别示于图/和表/6由图 /可见,:I/1(!,’ 上出现了#P1(!,’衍射峰6随 :. 的添加,载体#P1(!,’的衍射峰消失,:.的添加破坏 了部分#P1(!,’的晶形结构,使其以无定形态存在6 :I/:.(#)/1(!,’ 以及 :I/:.,! 氧化催化剂上存在 的主要结晶相为 :.,! 萤石相,其衍射峰强度随 :./ 1(比的增加逐渐增强6氧化催化剂上未出现 :I, 的衍射峰,这说明活性组分 :I可能主要以高度分 散的状态存在[/%] 6 由表/中的结果可见,少量 :.(:./1(B"F"/) 的添加显著降低了 :I/1(!,’ 催化剂的比表面积6 而当:.添加量进一步升高,催化剂比表面积先增大 后减小6当 :./1(比为 "F/% 时,比 表 面 积 达 到 最 大6:I/:.,! 具有最小的比表面积6:.添加对 :I/ 1(!,’ 比表面积的影响机理和变化规律还有待深入 研究6结合此后的活性数据发现,催化剂活性与比 表面积之间并没有线性关系,因此我们认为比表面 积并不是影响催化剂活性的主要因素6 表 ! $%/$&(!)/’()*# 系列氧化催化剂的比表面积 3>L(./ 8I2T>W.>2.>JTK0.JU)V>K)J*W>K>(XMKM JT:I/:.(#)/1(!,’ :>K>(XMK <93MI2T>W.>2.>(#!/$) :I/1(!,’ !!N6! :I/:.("6"/)/1(!,’ /QD6N :I/:.("6"E)/1(!,’ !’Q6E :I/:.("6/%)/1(!,’ ’!D6C :I/:.("6’")/1(!,’ /E"6E :I/:.,! D%6’ 为了研究 :.的添加使 :I/:.(#)/1(!,’ 系列 氧化催化剂活性提高的原因,下面进一步对 :I/:. (#)/1(!,’ 系列催化剂进行 -!P345及 748表征6 )"!") 1)203,表征结果 图 ! 是 :I/:.(#)/1(!,’ 催 化 剂 的 -!P345 谱,可见随着 :./1(比变化,催化剂的 -!P345 谱发 生了显著的变化6:I/1(!,’ 在!Q/&和’"!&出现 了两个还原峰,分别记为$和%6随着 :.的添加,$ 峰消失,%峰逐渐增大并向低温移动6当 :./1(比 大于"F"E时,#峰开始出现6:./1(比为"F’"时, 出现了新峰&6 %!E 催 化 学 报 第!N卷
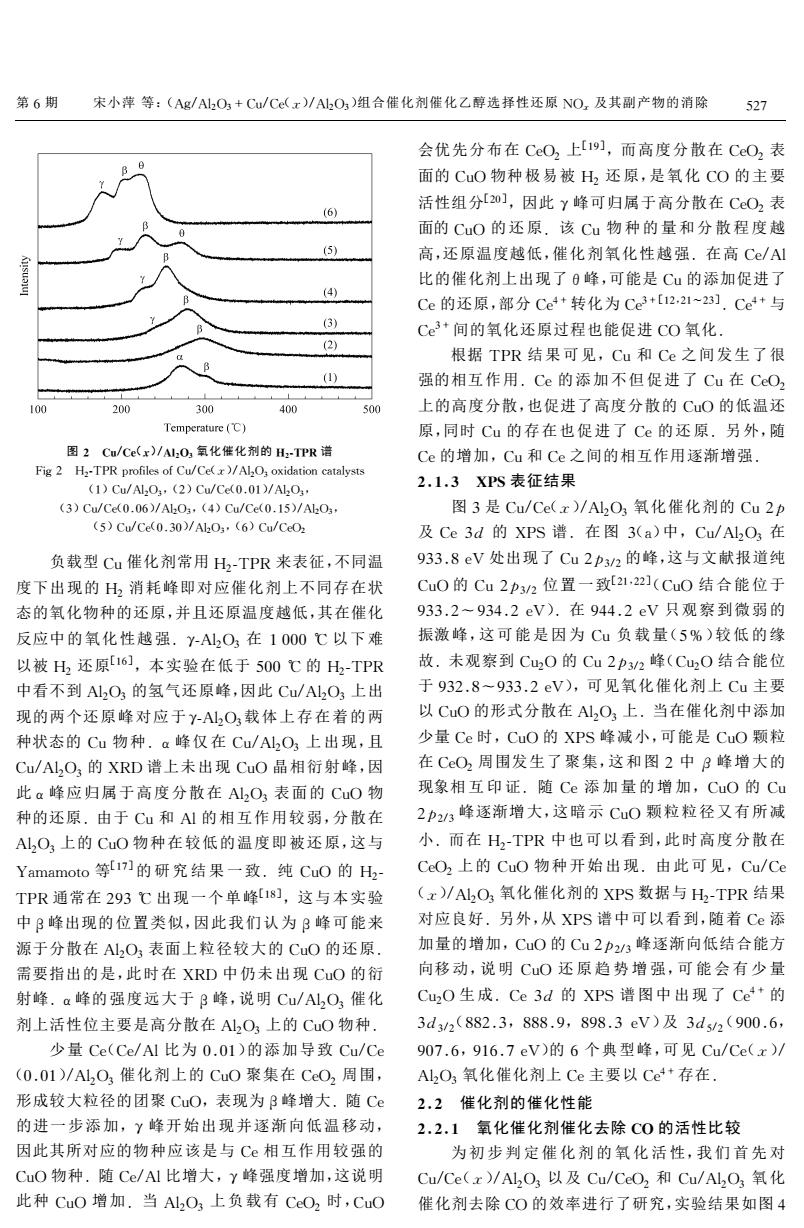
第6期 宋小萍等:(Ag/Al,O3+Cu/Ce(x)/Al2O3)组合催化剂催化乙醇选择性还原NO及其副产物的消除 527 会优先分布在CcO2上[191,而高度分散在CcO2表 面的CuO物种极易被H2还原,是氧化CO的主要 (6) 活性组分[20],因此y峰可归属于高分散在CeO2表 面的CuO的还原.该Cu物种的量和分散程度越 (5) 高,还原温度越低,催化剂氧化性越强.在高Ce/AI 比的催化剂上出现了0峰,可能是Cu的添加促进了 (4) Ce的还原,部分Ce4+转化为Ce3+12,21~23].Ce+与 (3) Ce3+间的氧化还原过程也能促进CO氧化. (2) 根据TPR结果可见,Cu和Ce之间发生了很 (1) 强的相互作用.Ce的添加不但促进了Cu在CeO2 100 200 300 400 500 上的高度分散,也促进了高度分散的CuO的低温还 Temperature (C) 原,同时Cu的存在也促进了Ce的还原.另外,随 图2Cu/Ce(x)/,O,氧化催化剂的H-TPR谱 Ce的增加,Cu和Ce之间的相互作用逐渐增强. Fig 2 H2-TPR profiles of Cu/Ce(x)/AlO oxidation catalysts (1)Cu/A2O3,(2)Cu/Ce(0.01)/Al2O3, 2.1.3XPS表征结果 (3)Cu/Ce(0.06)/A2O3,(4)Cu/Ce(0.15)/Ah2O3 图3是Cu/Ce(x)/Al2O3氧化催化剂的Cu2p (5)Cu/Ce(0.30)/Al2O3,(6)Cu/CeO2 及Ce3d的XPS谱.在图3(a)中,Cu/Al2O3在 负载型Cu催化剂常用H2-TPR来表征,不同温 933.8eV处出现了Cu2p3/2的峰,这与文献报道纯 度下出现的H2消耗峰即对应催化剂上不同存在状 CuO的Cu2p3/2位置一致2122](Cu0结合能位于 态的氧化物种的还原,并且还原温度越低,其在催化 933.2~934.2eV).在944.2eV只观察到微弱的 反应中的氧化性越强.Y-A12O3在1000℃以下难 振激峰,这可能是因为Cu负载量(5%)较低的缘 以被H2还原16],本实验在低于500℃的H2-TPR 故.未观察到Cu2O的Cu2p3/2峰(Cu2O结合能位 中看不到Al2O3的氢气还原峰,因此Cu/Al2O3上出 于932.8~933.2eV),可见氧化催化剂上Cu主要 现的两个还原峰对应于Y-A2O3载体上存在着的两 以CuO的形式分散在A1,O3上.当在催化剂中添加 种状态的Cu物种.a峰仅在Cu/Al2O3上出现,且 少量Ce时,CuO的XPS峰减小,可能是CuO颗粒 Cu/Al,O3的XRD谱上未出现CuO晶相衍射峰,因 在CeO2周围发生了聚集,这和图2中B峰增大的 此a峰应归属于高度分散在Al2O3表面的CuO物 现象相互印证.随Ce添加量的增加,CuO的Cu 种的还原.由于Cu和A1的相互作用较弱,分散在 2p23峰逐渐增大,这暗示Cu0颗粒粒径又有所减 Al,O3上的CuO物种在较低的温度即被还原,这与 小.而在H2-TPR中也可以看到,此时高度分散在 Yamamoto等17的研究结果一致.纯CuO的H2 CeO2上的CuO物种开始出现.由此可见,Cu/Ce TPR通常在293℃出现一个单峰18],这与本实验 (x)/Al2O3氧化催化剂的XPS数据与H2-TPR结果 中β峰出现的位置类似,因此我们认为3峰可能来 对应良好.另外,从XPS谱中可以看到,随着Ce添 源于分散在Al2O3表面上粒径较大的CuO的还原. 加量的增加,CuO的Cu2p2/3峰逐渐向低结合能方 需要指出的是,此时在XRD中仍未出现CuO的衍 向移动,说明CuO还原趋势增强,可能会有少量 射峰.a峰的强度远大于B峰,说明Cu/A,O,催化CuO生成.Ce3d的XPS谱图中出现了Ce+的 剂上活性位主要是高分散在Al2O3上的CuO物种. 3d3/2(882.3,888.9,898.3eV)及3d52(900.6, 少量Ce(Ce/Al比为0.01)的添加导致Cu/Ce 907.6,916.7eV)的6个典型峰,可见Cu/Ce(x)/ (0.01)/Al,O3催化剂上的CuO聚集在CeO2周围, Al2O3氧化催化剂上Ce主要以Ce4+存在. 形成较大粒径的团聚CuO,表现为B峰增大.随Ce 2.2催化剂的催化性能 的进一步添加,Y峰开始出现并逐渐向低温移动,2.2.1氧化催化剂催化去除C0的活性比较 因此其所对应的物种应该是与Ce相互作用较强的 为初步判定催化剂的氧化活性,我们首先对 CuO物种.随Ce/Al比增大,Y峰强度增加,这说明 Cu/Ce(x)/Al,O3以及Cu/CeO2和Cu/Al,O3氧化 此种CuO增加.当Al2O3上负载有CeO2时,CuO 催化剂去除CO的效率进行了研究,实验结果如图4
图 ! "#/"$(!)/%&!’( 氧化催化剂的 )!*+,-谱 !"#$ %$&’()*+,-"./0,-12/1/(!)/3.$45,6"789",:;898.<090 (=)12/3.$45,($)12/1/(>?>=)/3.$45, (5)12/1/(>?>@)/3.$45,(A)12/1/(>?=B)/3.$45, (B)12/1/(>?5>)/3.$45,(@)12/1/4$ 负载型12催化剂常用 %$&’()来表征,不同温 度下出现的 %$ 消耗峰即对应催化剂上不同存在状 态的氧化物种的还原,并且还原温度越低,其在催化 反应中的氧化性越强C!&3.$45 在 =>>> D以下难 以被 %$ 还原[=@],本实验在低于B>> D的 %$&’() 中看不到 3.$45 的氢气还原峰,因此 12/3.$45 上出 现的两个还原峰对应于!&3.$45载体上存在着的两 种状态的 12物种C"峰仅在 12/3.$45 上出现,且 12/3.$45 的 E)F谱上未出现 124 晶相衍射峰,因 此"峰应归属于高度分散在 3.$45 表面的 124 物 种的还原C由于 12和 3.的相互作用较弱,分散在 3.$45 上的 124物种在较低的温度即被还原,这与 G8H8H,9,等[=I]的 研 究 结 果 一 致C纯 124 的 %$& ’()通常在$J5 D出现一个单峰[=K],这与本实验 中#峰出现的位置类似,因此我们认为#峰可能来 源于分散在 3.$45 表面上粒径较大的 124 的还原C 需要指出的是,此时在 E)F 中仍未出现 124 的衍 射峰C"峰的强度远大于#峰,说明 12/3.$45 催化 剂上活性位主要是高分散在 3.$45 上的 124物种C 少量 1/(1//3.比为>?>=)的添加导致 12/1/ (>?>=)/3.$45 催化剂上的 124 聚集在 1/4$ 周围, 形成较大粒径的团聚 124,表现为#峰增大C随 1/ 的进一步添加,!峰开始出现并逐渐向低温移动, 因此其所对应的物种应该是与 1/相互作用较强的 124物种C随 1//3.比增大,!峰强度增加,这说明 此种 124 增加C当 3.$45 上负载有 1/4$ 时,124 会优先分布在 1/4$ 上[=J],而高度分散在 1/4$ 表 面的 124物种极易被 %$ 还原,是氧化 14 的主要 活性组分[$>],因此!峰可归属于高分散在 1/4$ 表 面的 124 的还原C该 12物 种 的 量 和 分 散 程 度 越 高,还原温度越低,催化剂氧化性越强C在高 1//3. 比的催化剂上出现了$峰,可能是 12的添加促进了 1/的还原,部分1/AL转化为1/5L[=$,$=%$5] C1/AL与 1/5L间的氧化还原过程也能促进 14氧化C 根据 ’() 结果可见,12和 1/之间发生了很 强的相互作用C1/的添加不但促进了 12在 1/4$ 上的高度分散,也促进了高度分散的 124 的低温还 原,同时 12的存在也促进了 1/的还原C另外,随 1/的增加,12和 1/之间的相互作用逐渐增强C !./.( 0,1表征结果 图5是 12/1/(!)/3.$45 氧化催化剂的 12$" 及 1/5# 的 E(M 谱C在 图 5(8)中,12/3.$45 在 J55?K/N 处出现了 12$"5/$的峰,这与文献报道纯 124的 12$"5/$ 位置一致[$=,$$(] 124 结合能位于 J55?$%J5A?$/N)C在JAA?$/N 只观察到微弱的 振激峰,这可能是因为 12负载量(BO)较 低 的 缘 故C未观察到 12$4 的 12$"5/$ 峰(12$4 结合能位 于J5$?K%J55?$/N),可见氧化催化剂上 12主要 以 124的形式分散在 3.$45 上C当在催化剂中添加 少量 1/时,124的 E(M峰减小,可能是 124 颗粒 在1/4$ 周围发生了聚集,这和图$中! 峰增大的 现象相 互 印 证C随 1/添 加 量 的 增 加,124 的 12 $"$/5峰逐渐增大,这暗示 124 颗粒粒径又有所减 小C而在 %$&’() 中也可以看到,此时高度分散在 1/4$ 上的 124 物种开始出现C由此可见,12/1/ (!)/3.$45 氧化催化剂的E(M数据与 %$&’()结果 对应良好C另外,从 E(M谱中可以看到,随着 1/添 加量的增加,124的12$"$/5峰逐渐向低结合能方 向移 动,说 明 124 还 原 趋 势 增 强,可 能 会 有 少 量 12$4 生成C1/5# 的 E(M 谱图中 出 现 了 1/AL 的 5#5/$ (KK$?5,KKK?J,KJK?5/N)及 5#B/$ (J>>?@, J>I?@,J=@?I/N)的@个典型峰,可见 12/1/(!)/ 3.$45 氧化催化剂上 1/主要以 1/AL存在C !.! 催化剂的催化性能 !.!./ 氧化催化剂催化去除 "’的活性比较 为 初 步 判 定 催 化 剂 的 氧 化 活 性,我 们 首 先 对 12/1/(!)/3.$45 以及 12/1/4$ 和 12/3.$45 氧化 催化剂去除14的效率进行了研究,实验结果如图A 第@期 宋小萍 等:(3#/3.$45L12/1/(!)/3.$45)组合催化剂催化乙醇选择性还原 P4! 及其副产物的消除 B$I
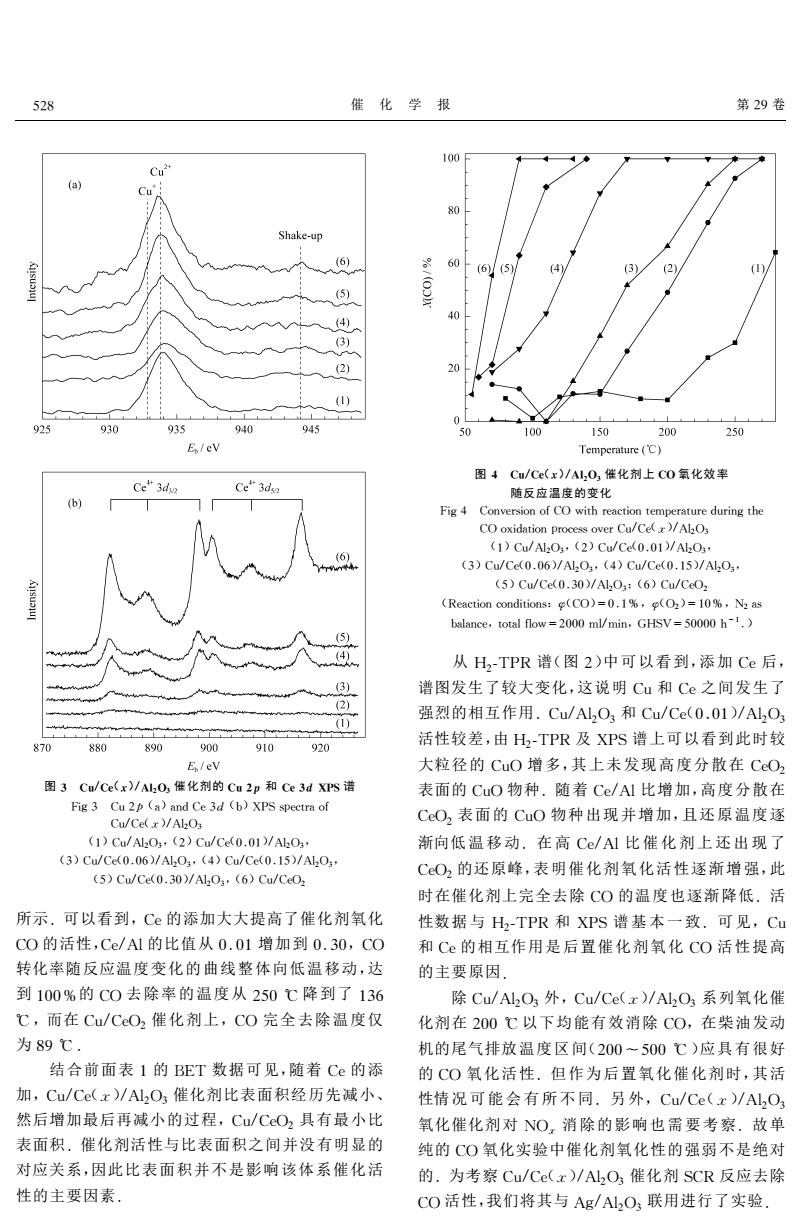
528 催化学报 第29卷 100 Cu 80 Shake-up 60(6)5) (4 (3) (2) (1) (5) (oD (4) 40 (3) (2) 20 (1) 0 925 930 935 940 945 0 100 150 200 250 E/ev Temperature (C) 图4Cu/Ce(x)/AlO3催化剂上C0氧化效率 Ce3du Ce"3ds 随反应温度的变化 Fig 4 Conversion of CO with reaction temperature during the CO oxidation process over Cu/Ce(/Al2O (1)Cu/Al2O3,(2)Cu/Ce(0.01)/Al2O3 (6) (3)Cu/Ce(0.06)/Al2O3,(4)Cu/Ce(0.15)/Al2O3 (5)Cu/Ce(0.30)/Al2O3:(6)Cu/CeO2 Reaction conditions:(CO)=0.1%,(2)=10%,N2 as balance,total flow=2000 ml/min.GHSV=50000 h-1.) (5) (4) 从H2-TPR谱(图2)中可以看到,添加Ce后, (3) 谱图发生了较大变化,这说明Cu和Ce之间发生了 (2) (1) 强烈的相互作用.Cu/AlO3和Cu/Ce(0.01)/AlO3 870 880 890900910 920 活性较差,由H2-TPR及XPS谱上可以看到此时较 E/ev 大粒径的CuO增多,其上未发现高度分散在CO2 图3Cu/Ce(x)/A,O3催化剂的Cu2p和Ce3dXPS谱 表面的CuO物种.随着Ce/Al比增加,高度分散在 Fig 3 Cu 2p (a)and Ce 3d (b)XPS spectra of Cu/Ce(x)/Al2O; CO2表面的CuO物种出现并增加,且还原温度逐 (1)Cu/Al2O3,(2)Cu/Ce(0.01)/Ah2O3, 渐向低温移动.在高Ce/Al比催化剂上还出现了 (3)Cu/Ce(0.06)/Al2O3,(4)Cu/Ce(0.15)/AlO3, CO2的还原峰,表明催化剂氧化活性逐渐增强,此 (5)Cu/Ce(0.30)/Al2O,(6)Cu/CeO2 时在催化剂上完全去除CO的温度也逐渐降低.活 所示.可以看到,Ce的添加大大提高了催化剂氧化 性数据与H2TPR和XPS谱基本一致.可见,Cu CO的活性,Ce/Al的比值从0.01增加到0.30,CO 和Ce的相互作用是后置催化剂氧化CO活性提高 转化率随反应温度变化的曲线整体向低温移动,达 的主要原因 到100%的C0去除率的温度从250℃降到了136 除Cu/Al2O3外,Cu/Ce(x)/Al2O3系列氧化催 ℃,而在Cu/CeO2催化剂上,CO完全去除温度仅 化剂在200℃以下均能有效消除CO,在柴油发动 为89℃. 机的尾气排放温度区间(200~500℃)应具有很好 结合前面表1的BET数据可见,随着Ce的添 的CO氧化活性.但作为后置氧化催化剂时,其活 加,Cu/Ce(x)/Al2O3催化剂比表面积经历先减小、 性情况可能会有所不同.另外,Cu/Ce(x)/AlO) 然后增加最后再减小的过程,Cu/CO2具有最小比 氧化催化剂对NO,消除的影响也需要考察.故单 表面积.催化剂活性与比表面积之间并没有明显的 纯的CO氧化实验中催化剂氧化性的强弱不是绝对 对应关系,因此比表面积并不是影响该体系催化活 的.为考察Cu/Ce(x)/Al2O3催化剂SCR反应去除 性的主要因素。 CO活性,我们将其与Ag/Al2O3联用进行了实验
图 ! "#/"$(!)/%&’(! 催化剂的 "#’" 和 "$!#)*+谱 !"#$ %&’!(()()*%+$"(,)-./01+234(56 %&/%+(#)/78’9$ (:)%&/78’9$,(’)%&/%+(;<;:)/78’9$, ($)%&/%+(;<;=)/78’9$,(>)%&/%+(;<:?)/78’9$, (?)%&/%+(;<$;)/78’9$,(=)%&/%+9’ 图 , "#/"$(!)/%&’(! 催化剂上 "(氧化效率 随反应温度的变化 !"#> %5)@+40"5)56%9A"3B4+(23"5)3+C1+4(3&4+*&4")#3B+ %95D"*(3"5)1452+005@+4%&/%+(#)/78’9$ (:)%&/78’9$,(’)%&/%+(;<;:)/78’9$, ($)%&/%+(;<;=)/78’9$,(>)%&/%+(;<:?)/78’9$, (?)%&/%+(;<$;)/78’9$;(=)%&/%+9’ (E+(23"5)25)*"3"5)0:! (%9)F;<:G,! (9’)F:;G,H’(0 ,(8()2+,353(8685AF’;;;C8/C"),IJ/KF?;;;;BL:M) 所示M可以看到,%+的添加大大提高了催化剂氧化 %9的活性,%+/78的比值从;M;:增加到;M$;,%9 转化率随反应温度变化的曲线整体向低温移动,达 到:;;G的 %9去除率的温度从’?; N降到了:$= N,而在 %&/%+9’ 催化剂上,%9 完全去除温度仅 为OP NM 结合前面表:的 QRS 数据可见,随着 %+的添 加,%&/%+(#)/78’9$ 催化剂比表面积经历先减小、 然后增加最后再减小的过程,%&/%+9’ 具有最小比 表面积M催化剂活性与比表面积之间并没有明显的 对应关系,因此比表面积并不是影响该体系催化活 性的主要因素M 从 J’TS.E 谱(图’)中可以看到,添加 %+后, 谱图发生了较大变化,这说明 %&和 %+之间发生了 强烈的相互作用M%&/78’9$ 和 %&/%+(;<;:)/78’9$ 活性较差,由 J’TS.E及 -./谱上可以看到此时较 大粒径的 %&9 增多,其上未发现高度分散在 %+9’ 表面的 %&9物种M随着 %+/78比增加,高度分散在 %+9’ 表面的 %&9 物种出现并增加,且还原温度逐 渐向低温移动M在高 %+/78比催 化 剂 上 还 出 现 了 %+9’ 的还原峰,表明催化剂氧化活性逐渐增强,此 时在催化剂上完全去除 %9 的温度也逐渐降低M活 性数据与 J’TS.E 和 -./谱基本一致M可见,%& 和 %+的相互作用是后置催化剂氧化 %9 活性提高 的主要原因M 除 %&/78’9$ 外,%&/%+(#)/78’9$ 系列氧化催 化剂在’;; N以下均能有效消除 %9,在柴油发动 机的尾气排放温度区间(’;;!?;; N)应具有很好 的 %9 氧化活性M但作为后置氧化催化剂时,其活 性情况可能会有所不 同M另 外,%&/%+(#)/78’9$ 氧化催化剂对 H9# 消除的影响也需要考察M故单 纯的 %9氧化实验中催化剂氧化性的强弱不是绝对 的M为考察 %&/%+(#)/78’9$ 催化剂/%E反应去除 %9活性,我们将其与 7#/78’9$ 联用进行了实验M ?’O 催 化 学 报 第’P卷